




la Newsletter
Bassa statura e sindromi rare | Il sospetto di una condizione...
Il sospetto di una condizione genetica di tipo sindromico nel...
La crescita è uno dei parametri di salute più evidenti ed è un fenomeno complesso in cui giocano ruoli importanti diversi fattori, sia genetici che ormonali, nutrizionali e ambientali. Il riscontro della bassa statura (short stature, SS), definita come deviazione della statura superiore a 2 DS al di sotto della media della popolazione di riferimento distinta per sesso o della statura target della famiglia, in un bambino è causa di preoccupazione per la famiglia e deve essere motivo di attenzione e quindi di accurata valutazione.
La Società Europea di Endocrinologia Pediatrica (ESPE) ha proposto nel 2007 una classificazione della bassa statura, aggiornata nel 2016, suddivisa in tre grandi categorie: bassa statura primaria, secondaria ed idiopatica. Nella prima categoria sono comprese le condizioni sindromiche su base genetica, il nato piccolo per età gestazionale con deficit di recupero di crescita e le displasie scheletriche, mentre nella seconda sono incluse condizioni dovute a cause ormonali, nutrizionali e ambientali o a malattie specifiche di organo. Nella terza le basse stature idiopatiche, quelle non associate a nessuna altra anomalia né organica né funzionale.
Negli ultimi anni il progresso tecnologico ha reso disponibili, anche in relazione alla riduzione notevole dei costi, approcci diagnostici più sofisticati come il sequenziamento di nuova generazione (NGS). La disponibilità di questo tipo di tecnica diagnostica ha facilitato la diagnosi di condizioni estremamente rare e ha permesso di identificare nuovi geni responsabili di nuove sindromi, di associare nuovi geni a condizioni sindromiche già note e di raggruppare diverse condizioni in base ai pathway molecolari implicati. I geni identificati, infatti, spesso appartengono a pathway già noti in cui sono coinvolti geni responsabili di altre condizioni sindromiche talora con quadro clinico in parte sovrapponibile, ad esempio il pathway SWI/SNF.
Il percorso diagnostico è sempre più complesso a causa della notevole eterogeneità sia clinica (malattie diverse dovute a mutazioni in domini diversi di uno stesso gene, con perdita o guadagno di funzione) che genetica, nello specifico allelica (stessa condizione dovuta a mutazioni diverse in uno stesso gene) o di locus (fenotipi simili da mutazioni in geni diversi). Il catalogo Online Mendelian Inheritance in Man (OMIM) nell’aprile 2019 riporta oltre 2000 condizioni con SS.
Nella review, suddivisa in due parti, ci focalizzeremo su alcune specifiche forme di bassa statura primaria come definita dall’ESPE. Questa prima parte è dedicata ad alcune condizioni sindromiche, la seconda alle displasie scheletriche. L’obiettivo è illustrare alcune condizioni rare già definite clinicamente ma di recente definizione etiopatogenetica e alcune nuove condizioni definite clinicamente in seguito ad analisi di dati genomici, il cosiddetto “Reverse Phenotyping”.
Sindromi genetiche
Il sospetto di una condizione genetica di tipo sindromico nel percorso diagnostico di un paziente con bassa statura nasce dall’evidenza di anomalie del fenotipo sia di tipo malformativo che neurologico/organico che debbono essere ricercate con grande attenzione. In prima istanza vengono prese in considerazione cause ben note di sindromi cromosomiche o di sindromi genomiche (da CNV microdel/microdup) o sindromi monogeniche più facilmente identificabili; alcuni esempi che rientrano in questa categoria: le sindromi di Down, di Turner, di Di George (22q11.2), di Cornelia de Lange, di Bloom, Kabuki, di Noonan, di Prader-Willi, di Rubinstein-Taybi, di Silver-Russel, di Williams. Di seguito sono descritte alcune condizioni, raggruppate dove possibile secondo criteri clinici utili per il sospetto diagnostico o per il pathway coinvolto o entrambi; le malattie descritte sono riportate con altre condizioni non descritte nel testo anche nella tabella 1.
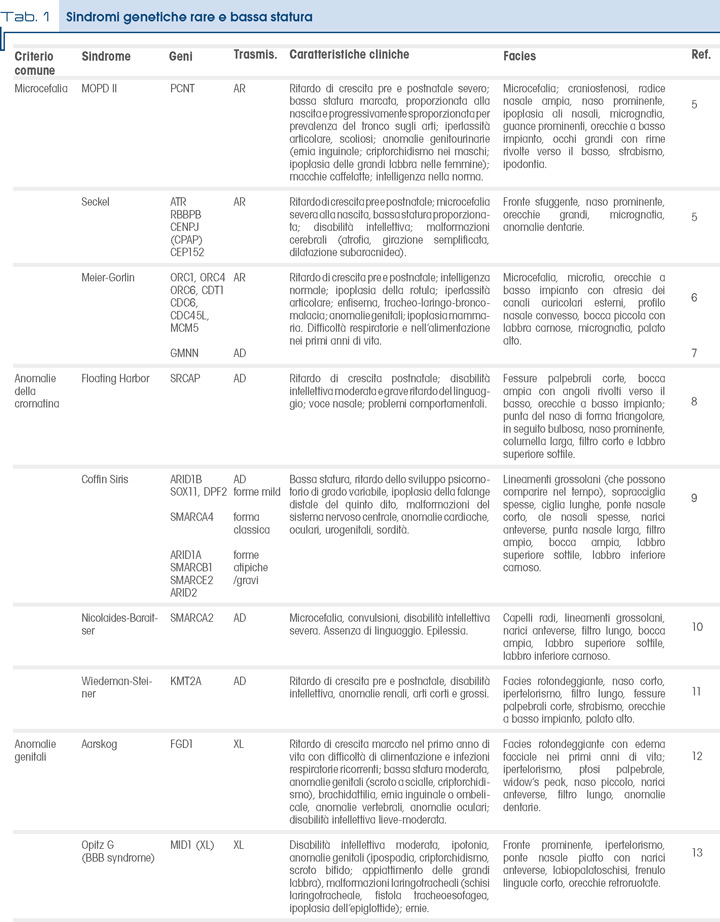
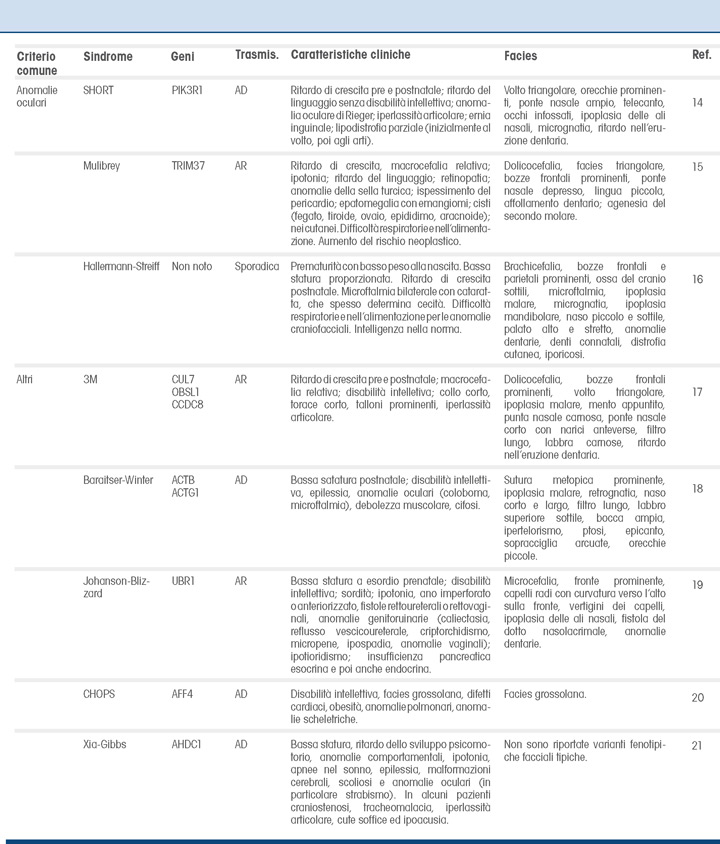
Bassa statura e microcefalia
La sindrome di Seckel (SekS) e la bassa statura primordiale con microcefalia di tipo II (Microcefalic Osteodisplastic Primordial Dwarfism, MOPD II) sono condizioni storicamente accomunate da un quadro clinico simile. Sono infatti caratterizzate da un ritardo di crescita sia pre che postnatale, molto più marcato nella MOPD II, e si differenziano principalmente per la disabilità intellettiva, presente nella SekS e solitamente assente nella MOPD II. Quest’ultima può manifestarsi con danno vascolare a livello del SNC con emorragia e infarto cerebrale nel 25% dei casi, causa di exitus nei primi anni di vita. La MOPD II è causata da alterazioni del gene PCNT, che codifica per la pericentrina, proteina chiave nella struttura del centrosoma e nella formazione del fuso mitotico, e la SekS è determinata da un’alterazione della risposta al danno del DNA mediata dalla proteina ATR, la quale interagisce con PCNT; gli altri geni responsabili della SeksS (CPAP, CEP152) fanno parte del medesimo pathway molecolare.
La sindrome di Meier-Gorlin (MGS) è caratterizzata da una triade: bassa statura, microtia e agenesia/ipoplasia della rotula a cui si aggiunge microcefalia. Presenta eterogeneità genetica ma i diversi geni causativi (ORC1, ORC4, ORC6, CDT1, CDC6, GMNN, CDC45 e MCM5) sono tutti coinvolti nello stesso pathway. I primi tre fanno parte del complesso ORC (origin recognition complex del DNA), il quale, dopo aver reclutato altre proteine, tra cui CDT1 e CDC6, attiva l’elicasi MCM5, che ha un ruolo fondamentale nella replicazione del DNA; per tutti questi geni l’ereditarietà è di tipo autosomico recessivo. Le mutazioni del gene GMNN, regolatore della trascrizione che interagisce con CDT1, causano un guadagno di funzione e la trasmissione è di tipo autosomico dominante.
Bassa statura da anomalie della cromatina
La sindrome Floating-Harbor presenta caratteristiche facciali tipiche, voce nasale, ritardo di crescita postnatale, disabilità intellettiva moderata con grave ritardo del linguaggio e problemi comportamentali. La trasmissione è di tipo autosomico dominante con alterazioni del gene SRCAP, che codifica per una proteina che è coinvolta nel rimodellamento della cromatina e interagisce con CREBBP, la cui alterazione è responsabile della sindrome di Rubinstein-Taybi (RTS). Mutazioni del gene CREBBP negli esoni 30 o 31 sono responsabili di una condizione con SS ma fenotipo diverso dalla RTS nota come sindrome di Menke-Hennekam.
La sindrome di Coffin-Siris (CSS) e la sindrome di Nicolaides-Baraitser (NBS) sono malattie clinicamente e geneticamente eterogenee. Tali condizioni possono essere causate da una mutazione in eterozigosi o da un riarrangiamento genomico in diversi geni, tutti codificanti per proteine che compongono un complesso proteico noto come SWI/SNF (Switch/Sucrose Non-Fermenting o anche BAF complex) coinvolto nel rimodellamento della cromatina e nella regolazione dell'espressione genica durante lo sviluppo. Gli elementi clinici dei pazienti con mutazioni nei geni che codificano per proteine che fanno parte di questo complesso (ARID1B, ARID1A, SMARCA4, SMARCB1, SMARCE1, SOX11, ARID2, DPF2, SMARCA2, PHF6) sono variabili, costituiscono un continuum clinico di cui la forma più severa è la NBS e differiscono per gravità e per alcune caratteristiche, a seconda del gene coinvolto. Le manifestazioni cliniche principali della CSS classica (mutazioni del gene SMARCA4) sono il ritardo dello sviluppo psicomotorio (di grado variabile), la bassa statura, l’ipoplasia della falange distale del quinto dito e una facies tipica. Possono essere presenti anche malformazioni del sistema nervoso centrale, anomalie cardiache, oculari, urogenitali, sordità e altri elementi clinici. Le caratteristiche facciali comprendono lineamenti grossolani (che possono comparire nel tempo), sopracciglia spesse, ciglia lunghe, ponte nasale corto, ali nasali spesse, narici anteverse, punta nasale larga, filtro ampio, bocca ampia, labbro superiore sottile, labbro inferiore carnoso. La NBS è dovuta a mutazioni del gene SMARCA2 presenta un fenotipo facciale tipico e un quadro clinico severo con microcefalia, convulsioni e disabilità intellettiva grave con assenza di linguaggio.
Bassa statura e anomalie dei genitali
La sindrome di Aarskog presenta bassa statura moderata con ritardo di crescita marcato nel primo anno di vita, legato a difficoltà di alimentazione e infezioni respiratorie ricorrenti; tipiche di questa condizione sono la facies e le anomalie genitali (scroto a scialle, criptorchidismo). L’ereditarietà è di tipo X-linked con mutazioni del gene FGD1, espresso a livello del citoplasma e del Golgi e coinvolto nell’organizzazione del citoscheletro.
La sindrome di Opitz G (BBB syndrome) presenta una facies caratteristica, labiopalatoschisi, disabilità intellettiva moderata, ipotonia, anomalie genitali e malformazioni laringotracheali. L’ereditarietà è di tipo X-linked con mutazioni nel gene MID1, una ubiquitina-E3-ligasi ancorata ai microtubuli che regola la degradazione proteica.
Bassa statura e anomalie oculari
La sindrome SHORT (Short stature, Hyperextensibility of joints, inguinal Hernia, Ocular depression, Rieger abnormality, Teething delay) è caratterizzata da ritardo di crescita pre e postnatale, iperlassità articolare, ernia inguinale, occhi infossati, anomalia oculare di Rieger (disgenesia del segmento anteriore) e ritardo della dentizione; è descritto un ritardo nello sviluppo del linguaggio senza disabilità intellettiva ed è tipica una lipodistrofia parziale che nel corso del tempo si estende dal volto agli arti e intolleranza glucidica con insorgenza precoce di diabete. La trasmissione è di tipo autosomico dominante ed è dovuta a varianti nel gene PIK3R1, che ha un ruolo chiave nei meccanismi della crescita cellulare.
La sindrome Mulibrey (MUscle, LIver, BRain, EYe) presenta un ritardo di crescita ad esordio prenatale con macrocefalia relativa, ipotonia muscolare, epatomegalia con emangiomi, lieve ritardo del linguaggio con abilità intellettive normali, anomalie della sella turcica e retinopatia; sono descritti un ispessimento del pericardio, la formazione di cisti (fegato, tiroide, ovaio, epididimo, aracnoide) e di nei cutanei ed un aumento del rischio neoplastico; la scarsa crescita deriva anche da difficoltà respiratorie e nell’alimentazione. La sindrome è dovuta a mutazioni nel gene TRIM37, una ubiquitina-E3-ligasi che regola la degradazione proteica, e presenta una trasmissione di tipo autosomico recessivo.
La sindrome di Hallermann-Streiff ha un fenotipo peculiare facilmente identificabile, caratterizzato da microftalmia bilaterale con cataratta ed ipoplasia mandibolare; i pazienti presentano prematurità con basso peso alla nascita e ritardo di crescita postnatale, legato anche alle difficoltà respiratorie e dell’alimentazione a causa delle anomalie craniofacciali; non è descritta disabilità intellettiva. Ad oggi non è stata identificata una causa molecolare.
Altre sindromi
La sindrome 3M (Miller, McKusick and Malvaux) è caratterizzata da una facies tipica, ritardo di crescita pre e postnatale, macrocefalia relativa, iperlassità articolare e disabilità intellettiva. La trasmissione è di tipo autosomico recessivo con alterazioni dei geni CUL7, OBSL1 e CCDC8, che codificano per proteine che compongono un complesso centrosomico necessario per la formazione dei microtubuli, l’integrità genomica e la crescita cellulare.
La sindrome di Baraitser-Winter o cerebrofrontofaciale, è caratterizzata da bassa statura postnatale, disabilità intellettiva, facies tipica, anomalie oculari e muscoloscheletriche. La trasmissione è di tipo autosomico dominante con mutazioni nei geni ACTB e ACTG1, che codificano per due isoforme dell’actina espresse in diversi tipi cellulari come componenti del citoscheletro e mediatori della motilità intracellulare; sono coinvolte inoltre nell’assemblaggio del sarcomero.
La sindrome di Johanson-Blizzard è caratterizzata da bassa statura a esordio prenatale, disabilità intellettiva e ipotonia, con una facies caratteristica; i pazienti presentano in misura variabile altre caratteristiche, quali anomalie ano-rettali e genitourinarie, sordità e ipotonia; sono frequenti l’ipotiroidismo e l’insufficienza pancreatica (sia esocrina che endocrina). La trasmissione è di tipo autosomico recessivo e la condizione è legata ad alterazioni del gene UBR1, che codifica per una componente del complesso ubiquitin-proetin-ligasi-E3.
La sindrome CHOPS (Cognitive impairment and Coarse facies, Heart defects, Obesity, Pulmonary involvement, Short stature and Skeletal dysplasia) presenta bassa statura, disabilità intellettiva, facies grossolana, difetti cardiaci, obesità, anomalie polmonari e anomalie scheletriche. La trasmissione è di tipo autosomico dominante con mutazioni nel gene AFF4 che determinano anomalie nell’elongazione legata alla RNA-polimerasi e quindi nei processi di trascrizione del DNA; negli stessi meccanismi sono coinvolte le coesine, le cui mutazioni sono responsabili di altre condizioni sindromiche note come “coesinopatie”, di cui fa parte la sindrome di Cornelia de Lange da considerare nella diagnosi differenziale.
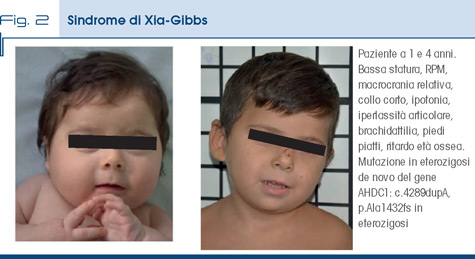 La sindrome di Xia-Gibbs (Fig. 2) è una malattia a trasmissione autosomica dominante dovuta a mutazioni in eterozigosi del gene AHDC1, che presenta domini di legame con il DNA ed è coinvolto nel rimodellamento della cromatina. Clinicamente è caratterizzata da ritardo globale dello sviluppo psicomotorio, anomalie comportamentali, ipotonia, apnee nel sonno, epilessia e malformazioni cerebrali; sono descritti anche ritardo di crescita, scoliosi e anomalie oculari (in particolare strabismo); non sono riportate varianti fenotipiche facciali tipiche della condizione. In alcuni pazienti sono riportati craniostenosi, tracheomalacia, iperlassità articolare, cute soffice ed ipoacusia. Alcuni elementi clinici sono presenti alla nascita, mentre altri compaiono nel tempo.
La sindrome di Xia-Gibbs (Fig. 2) è una malattia a trasmissione autosomica dominante dovuta a mutazioni in eterozigosi del gene AHDC1, che presenta domini di legame con il DNA ed è coinvolto nel rimodellamento della cromatina. Clinicamente è caratterizzata da ritardo globale dello sviluppo psicomotorio, anomalie comportamentali, ipotonia, apnee nel sonno, epilessia e malformazioni cerebrali; sono descritti anche ritardo di crescita, scoliosi e anomalie oculari (in particolare strabismo); non sono riportate varianti fenotipiche facciali tipiche della condizione. In alcuni pazienti sono riportati craniostenosi, tracheomalacia, iperlassità articolare, cute soffice ed ipoacusia. Alcuni elementi clinici sono presenti alla nascita, mentre altri compaiono nel tempo.
In questa review abbiamo presentato una breve panoramica su alcune malattie di più difficile identificazione al fine di sottolineare quanto sia importante giungere ad una diagnosi per conoscere la storia naturale e quindi la evoluzione della condizione.
La seconda parte della review, dedicata alle displasie scheletriche, verrà pubblicata sul n.2 di MR.
Bibliografia
- Wei C and Gregory JW. Physiology of normal growth. Paediatrics and Child Health 2009, 19:236-240
- Wit JM, Ranke MB, Kelnar CJH. ESPE classification of Paediatric Endocrine diagnosis Short Stature. Horm Res 2007;68(supp 2):1–5
- The International Classification of Pediatric Endocrine Diagnose (ICPED): www.icped.org/revisions/0/2015/diagnoses/
- Baron J, Sävendahl L, De Luca F et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol 2015; 11: 735–746.
- Piane M, Della Monica M, Piatelli G et al. Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II) Syndrome Previously Diagnosed as Seckel Syndrome: Report of a Novel Mutation of the PCNT Gene. Am J Med Genet 2009, 149A:2452-56.
- Vetro A, Savasta S, Russo Raucci A et al. MCM5: a new actor in the link between DNA replication and Meier-Gorlin syndrome. Eur J Hum Genet 2017, 25, 646-50
- Ting CY1, Bhatia NS2, Lim JY et al. Further delineation of CDC45-related Meier-Gorlin syndrome with craniosynostosis and review of literature. Eur J Med Genet. 2019 Apr 13. doi: 10.1016/j.ejmg.2019.04.009.
- Nikkel SM, Dauber A, de Munnik S et al. The phenotype of Floating-Harbor syndrome: clinical characterization of 52 individuals with mutations in exon 34 of SRCAP. OJRD 2013, 8:63
- Bögershausen N and Wollnik B. Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Front Mol Neurosci 2018, 11:252
- Vasileiou G, Vergarajauregui S, Endele S et al. - Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome. AM J Hum Genet 2018; 102:468-479
- Aggarwal A, Rodriguez-Buritica DF, Northrup H. Wiedemann-Steiner syndrome: Novel pathogenic variant and review of literature. Eur J Med Genet 2017; 60:285-288.
- Orrico A, Galli L, Faivre L et al. Aarskog–Scott Syndrome: Clinical Update and Report of Nine Novel Mutations of the FGD1 Gene. Am J Med Genet 2010, 152A:313–318.
- Meroni G. X-Linked Opitz G/BBB Syndrome. In: Adam MP, Ardinger HH, Pagon RA et al. editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993-2019.
- Avila M, Dyment DA, Sagen JV et al. Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: toward recommendation for molecular testing and management. Clin Genet 2016; 89: 501-06.
- Brigant B, Metzinger-Le Meuth V, Rochette J. TRIMming down to TRIM37: Relevance to Inflammation, Cardiovascular Disorders, and Cancer in MULIBREY Nanism. Int J Mol Sci 2018 Dec 24: E67.
- Schmidt J, Wollnik B. Hallermann– Streiff syndrome: A missing molecular link for a highly recognizable syndrome. Am J Med Genet 2018;178C:398–406.
- Simsek-Kiper PO, Taskiran E, Kosukcu C et al. Further expanding the mutational spectrum and investigation of genotype-phenotype correlation in 3M syndrome. Am J Med Genet 2019, 179A:1157-72.
- Verloes A, Drunat S, Pilz D, Di Donato N.et al. Baraitser-Winter Cerebrofrontofacial Syndrome. In: Adam MP, Ardinger HH, Pagon RA et al. editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993-2019.
- Sukalo M, Schäflein E, Schanze Iet al. Mutations in the human UBR1 gene and the associated phenotypic spectrum. Hum Mutat 2014;35:521-31.
- Raible SE, Mehta D, Bettale C |et al. Clinical and molecular spectrum of CHOPS syndrome. Am J Med Genet 2019; 179A:1126-1138.
- Jiang Y, Wangler MF, McGuire AL et al. The phenotypic spectrum of Xia-Gibbs syndrome. Am J Med Genet 2018;176A:1315-1326.