




la Newsletter
Bassa statura e sindromi rare: le displasie scheletriche | Le...
Le displasie scheletriche costituiscono un gruppo ampio ed...
La bassa statura è uno degli elementi clinici per cui più frequentemente un paziente viene sottoposto all’attenzione del pediatra. La Società Europea di Endocrinologia Pediatrica (ESPE) ha proposto nel 2007 una classificazione della bassa statura, aggiornata nel 2016, che la suddivide in tre grandi categorie: bassa statura primaria, secondaria ed idiopatica. Nella prima categoria sono comprese le condizioni sindromiche su base genetica (oggetto della prima parte di questa review, MR n.1/2020), il nato piccolo per età gestazionale con deficit di recupero di crescita e le displasie scheletriche sono oggetto di questa seconda parte.
Le displasie scheletriche costituiscono un gruppo ampio ed estremamente eterogeneo di anomalie di crescita e funzione del tessuto osteocartilagineo. La maggior parte è causa di bassa statura disarmonica/sproporzionata. L’espressività clinica varia dalla letalità perinatale in alcune condizioni fino al lieve ritardo di crescita in altre. La caratterizzazione clinica-radiologica è relativamente semplice ma la definizione pato-molecolare molto complessa.
Nell’ultima revisione del 2019 del gruppo di esperti dell’International Skeletal Dysplasias Society (ISDS), sono classificate 461 condizioni in 42 gruppi con criteri clinici, radiografici e/o molecolari. Sono riportati 437 geni causativi di 425 condizioni, pari al 92%. È importante rammentare che la diagnosi è possibile alla nascita o in utero soltanto per il 40% delle condizioni note.
La prevalenza varia molto nei diversi studi (da 2.1 a 4.7/10000 nati) a seconda dell’inserimento dei dati sui nati morti e sulle interruzioni di gravidanza. In ogni caso è accettabile una stima pari a 2 casi su 10.000 nati vivi e 20 casi su 10.000 nati morti.
Si rimanda a testi di anatomia e istologia per quanto riguarda la struttura del tessuto osseo ma è necessario comunque ricordare brevemente i due processi di sviluppo del tessuto osseo: l’ossificazione membranosa o diretta delle ossa della volta cranica e delle clavicole in cui le cellule progenitrici mesenchimali si differenziano via preosteoblasti in osteoblasti e l’ossificazione encondrale (cartilaginea) o indiretta delle ossa lunghe, vertebre e costole, dove le cellule mesenchimali progenitrici si differenziano in cellule pericondriali e condrociti. Il tessuto osseo è dinamico: subisce un continuo e regolare processo di neoformazione e riassorbimento grazie all’equilibrio funzionale di due linee cellulari osteoblasti e osteoclasti.
Un cenno al modello indiretto di ossificazione encondrale: alle estremità delle ossa lunghe abbiamo la cartilagine di accrescimento o Growth Plate (GP) dove si distinguono 4 zone cellulari: zona di riserva, zona di proliferazione, zona preipertrofica e ipertrofica in cui si completa il modello cartilagineo che subirà l’apposizione di tessuto osseo e la sua sostituzione con l’invasione contemporanea dei vasi sanguigni. La crescita in senso latero-laterale è dovuta alle cellule pericondriali che diverrano cellule del periostio nell’osso maturo.
Pathways di segnale
.jpg) Nello sviluppo del GP sono coinvolti numerosi pathways di segnale: Indian Hedgehog e PTHrP, BMPs, WNTs, Notch, CNP/NPR2 e FGFs. Tutti agiscono favorendo lo sviluppo della zona di proliferazione che comporta un allungamento finale dell’osso e inibiscono la differenziazione e maturazione dei condrociti (zone pre e ipertrofica) ad eccezione del pathway FGFs che ha un’azione diametralmente opposta (Fig. 1).
Nello sviluppo del GP sono coinvolti numerosi pathways di segnale: Indian Hedgehog e PTHrP, BMPs, WNTs, Notch, CNP/NPR2 e FGFs. Tutti agiscono favorendo lo sviluppo della zona di proliferazione che comporta un allungamento finale dell’osso e inibiscono la differenziazione e maturazione dei condrociti (zone pre e ipertrofica) ad eccezione del pathway FGFs che ha un’azione diametralmente opposta (Fig. 1).
Sono disponibili in letteratura eccellenti review sullo specifico tema a cui si rimanda.
Le tabelle riportano alcuni esempi, distinti da un punto di vista pato-molecolare in due sottogruppi:
- condizioni dovute a anomalie di geni in pathways di segnale coinvolti nello sviluppo del GP (Tab. 1);
- condizioni causate da anomalie dei geni coinvolti nella sintesi delle proteine della matrice extracellulare (Tab. 2).
Solo alcune delle condizioni riportate nella tabella sono descritte.
.jpg)
.jpg)
Pathway FGFs/FGFR
I fattori di crescita dei fibroblasti (FGF) sono alcune decine ma i recettori (FGFR), proteine integrali di membrana, soltanto quattro. Il recettore FGFR3 ha un ruolo fondamentale nello sviluppo del GP: agisce come modulatore negativo della condrogenesi inibendo la proliferazione dei condrociti. Mutazioni attivanti di questo gene comportano una riduzione dell’accrescimento a livello del GP, causa di un gruppo di condizioni, di gravità differente, diverse a seconda del domain della proteina modificato dalla mutazione.
La displasia tanatofora, letale in epoca neonatale, è una delle forme più gravi legate a mutazione in domain diversi del gene escluso il domain transmembrana. E’ caratterizzata da micromelia, coste corte, torace stretto, platispondilia: nel tipo 1 sono presenti femori curvi con aspetto definito a cornetta di telefono, mentre nel tipo 2 è presente il cranio a trifoglio.
L’acondroplasia è la displasia scheletrica più frequente causa di bassa statura disarmonica.
Ha una trasmissione di tipo autosomico dominante ma in oltre il 90% è dovuta a una mutazione de novo, che è sempre la stessa: la sostituzione di una glicina con arginina in posizione 380 della proteina (p.G380R) nel segmento transmembrana; sono riportate raramente altre due mutazioni. La ricorrenza dovuta a mosaicismo gonadico è eccezionale. Il fenotipo clinico è caratterizzato da bassa statura disarmonica per prevalenza del tronco sugli arti, brachidattilia con isodattilia, facies caratteristica con macrocefalia relativa, bozze frontali prominenti e radice nasale insellata, iperlordosi lombare e intelligenza normale. Segni radiologici peculiari sono: ossa tubulari corte e tozze, riduzione della distanza interpeduncolare in sede lombare, orizzontalizzazione dell’acetabolo e alterazioni metafisarie; alla nascita è tipico l’aspetto radiologico della pelvi a tridente e l’aspetto ovoidale radiotrasparente dell’estremità superiore del femore.
I pazienti possono andare incontro ad apnee notturne, che possono essere legate ad ipertrofia adenotonsillare oppure a cause centrali da compromissione dei centri del respiro per stenosi del forame magno. E’ importante il controllo del peso corporeo, perché questi pazienti hanno una maggiore tendenza all’obesità. Una mutazione specifica nel secondo domain intracellulare del recettore FGFR3, la sostituzione di una lisina in posizione 650 della proteina con una metionina (p.K650M), è responsabile di una forma particolare di acondroplasia con acanthosis nigricans e disabilità intellettiva (SADDAN). La posizione 650 è un Hotspot: sostituzioni della lisina 650 con altri aminoacidi sono causa di ipocondroplasia e displasia tanatofora.
L’ipocondroplasia è clinicamente caratterizzata da bassa statura non grave con disarmonia tra tronco e arti per prevalenza del tronco e alcuni segni radiologici peculiari ma non evidenti alla nascita: fibula distalmente più lunga della tibia e corpi vertebrali quadrangolari. Il quadro clinico alla nascita, a differenza dell’acondroplasia, non è immediatamente suggestivo della diagnosi, ma un’accurata valutazione clinica, anamnestica e radiologica permette di porre il sospetto.
Pathway WNT
I geni WNT sono responsabili di meccanismi di trasduzione del segnale e sono coinvolti in numerosi processi di sviluppo embrionale e proliferazione cellulare. Il pathway WNT è un induttore della linea osteoblastica e un inibitore della condrogenesi e della osteoclastogenesi e quindi del riassorbimento osseo. Mutazioni dei geni coinvolti nel pathway di WNT a livello del GP sono responsabili della sindrome di Robinow caratterizzata da bassa statura ad esordio postnatale, brevità acro-mesomelica degli arti e anomalie genitali con difficoltà di attribuzione del sesso nei maschi. Sono note una forma dominante, legata ai geni WNT5A, DVL1, DVL3 con anomalie scheletriche meno gravi e anomalie dentarie più importanti rispetto alla forma recessiva, causata da mutazioni nel gene ROR2. I geni responsabili sono tutti coinvolti negli stessi meccanismi molecolari; WNT5A, gene critico per i processi di sviluppo che richiedono la migrazione cellulare, è un corecettore di ROR2, fondamentale per lo sviluppo embrionale, mentre DVL1 e DVL3 sono mediatori a valle nello stesso pathway.
Pathway PTHrP/IHH
Le proteine PTHrP (peptide relato al paratormone) e IHH (Indian Hedgehog) determinano nel GP un feedback negativo che controlla la proliferazione e inibisce l’ipertrofia e la maturazione dei condrociti. Mutazioni nei geni coinvolti in questo pathway causano quadri clinici diversi. La displasia metafisaria tipo Jansen è una condizione a trasmissione autosomica dominante legata a mutazioni del gene PTHR1, recettore del paratormone. È caratterizzata da bassa statura disarmonica grave, arti corti e curvi, intelligenza normale, faccia prominente con microretrognatia, palato alto ed ipertelorismo, ipercalcemia ed ipofosforemia. La bassa statura, presente già alla nascita, diventa maggiormente evidente negli anni successivi, con marcata sproporzione tra arti e tronco. In età adulta possono comparire iperplasia frontonasale e sovraorbitaria, contratture articolari e sclerosi di alcune ossa craniche con compressione dei nervi cranici e deficit visivi e/o uditivi. La gravità del quadro clinico è variabile in rapporto alla localizzazione della specifica mutazione.
Pathways CNP/NPR2
La displasia acromesomelica tipo Maroteuax (AMDM) è una malattia a trasmissione autosomica recessiva caratterizzata da bassa statura disarmonica, normale intelligenza, mani con dita corte e tozze, cute delle mani ridondante e lassa e piedi con dita corte ma alluce largo. Caratteristico aspetto alla radiografia delle mani è la brevità di tutti i segmenti: metacarpi corti, curvi e tozzi, falangi corte e tozze “bullet-like”. Il gene responsabile, NPR2, codifica per il recettore tipo B del CNP, Peptide natriuretico tipo C, che è il suo specifico ligando. Si tratta di uno dei tre recettori dei tre peptidi natriuretici: ANP (atrial), BNP (brain) e CNP. Il recettore è un omodimero guanilico-ciclasi che converte GTP in cGMP. La cascata CNP/NPR2 agisce in maniera autocrina/paracrina come regolatore positivo dell’ossificazione encondrale stimolando la proliferazione dei condrociti e la sintesi di proteine della matrice extracellulare. Mutazioni inattivanti del gene sono causa di perdita di funzione e responsabili della AMDM. Mutazioni in eterozigosi di NPR2 causano bassa statura idiopatica senza anomalie scheletriche e mutazioni con guadagno di funzione una overespressione di CNP che causa una sindrome con overgrowth e alta statura.
Difetti delle proteine della matrice Extracellulare
La matrice extracellulare è una struttura dinamica ricca di proteine multimodulari altamente glicosilate e di polisaccaridi che sorreggono meccanicamente e orientano le cellule modulando le loro funzioni fisiologiche. Sono cinque le classi di macromolecole: collageni, elastina, acido ialuronico, proteoglicani, glicoproteine.
Aggrecano
È una proteina della matrice extracellulare codificata dal gene ACAN che ha un ruolo strutturale e funzionale fondamentale nella cartilagine di accrescimento. Costituisce il “core” di un proteoglicano della matrice extracellulare a cui sono legate 100 catene di glicosaminogicani tipo Condroitinsolfato e 30 tipo Cheratosolfato.
Varianti patogenetiche del gene ACAN sono causa di diverse condizioni: una forma autosomica dominante di displasia spondiloepifisaria, tipo Kimberley, una displasia spondiloepimetafisaria autosomica recessiva, una displasia epifisaria multipla con macrocefalia e facies peculiare, autosomica recessiva, una forma autosomica recessiva di ostecondrite dissecante familiare e anche casi di bassa statura idiopatica autosomica dominante.
Collageni
La famiglia di proteine più abbondanti della matrice è rappresentata dal collagene di cui sono stati identificati 28 differenti tipi, coinvolti nella formazione di un network di fibrille e microfibrille, membrane basali e altre strutture della matrice extracellulare.
Collagene tipo 1 ha una struttura trimerica costituita da due proteine A1 (1464 aminoacidi) una A2 (1366 aminoacidi) codificate rispettivamente da due geni COL1A1 e COL1A2. La struttura ha una conformazione a tripla elica: ogni proteina mostra una sequenza che presenta una glicina ogni tre aminoacidi. La biosintesi è un processo molto complesso in quanto la struttura subisce un processo di maturazione con modifiche post-trascrizionali sia intra che extracellulari. Il Collagene di tipo 1 rappresenta il 90% della massa organica dell’osso e tendini ed è il tipo maggiormente presente nella cute, legamenti e cornea. Mutazioni in uno dei due geni sono causa di anomalie quantitative con ridotta sintesi o qualitative da alterazioni del processo di maturazione post-trascrizionale causa principale dell’osteogenesi imperfetta. Si tratta di una condizione caratterizzata da fragilità ossea, osteopenia, deformità scheletriche e ritardo di crescita con prevalenza di 1 caso su 10.000 nati. Clinicamente si distinguono 4 fenotipi (Tab. 2): tipo I lieve da difetti quantitativi mentre difetti qualitativi sono causa del tipo II letale in epoca neonatale, del tipo III deformante e del tipo IV moderato. Mutazioni dei due geni COL1 sono la causa del 90% dei casi e si trasmettono con modalità autosomica dominante. Il 10% di fenotipi II, III e IV sono dovuti ad alterazioni degli altri 14 geni finora identificati, tutti a ereditarietà autosomica recessiva tranne uno a ereditarietà X–linked. Va ricordata la possibilità relativamente rara di casi di mosaicismo gonadico. Infine di recente è stato identificato un fenotipo particolare tipo V che oltre alla triade classica di segni ne presenta alcuni clinico-radiografici specifici (Tab. 2) e si trasmette con modalità autosomica dominante.
Collagene tipo 9 ha una struttura eterotrimerica a conformazione a tripla elica costituita da tre proteine codificate da tre geni COL9A1, COL9A2 e COL9A3. Mutazioni in eterozigosi sono causa di displasia epifisaria multipla (Tab. 2). Si tratta di una condizione con prevalenza di 1/10.000- 1/20.000 persone. E’ una condizione geneticamente eterogenea ad ereditarietà autosomica dominante da mutazioni in uno dei tre geni del COL9, o del gene COMP (Cartilage oligomeric matrix protein) o del gene MAT-3 (matrilina-3) o autosomica recessiva da mutazioni del gene SLC26A2 (trasportatore del solfato) o dal gene CANT1 (Calcium activated nucleotidase 1) che è una proteina del reticolo endoplasmatico/apparato di Golgi critica per l’ossificazione encodrale e la biosintesi dei glicosaminoglicani.
La malattia ha un esordio precoce interessando tutte le epifisi fino alla distruzione della cartilagine articolare.
In figura 2 alcuni esempi di anomalie radiografiche.
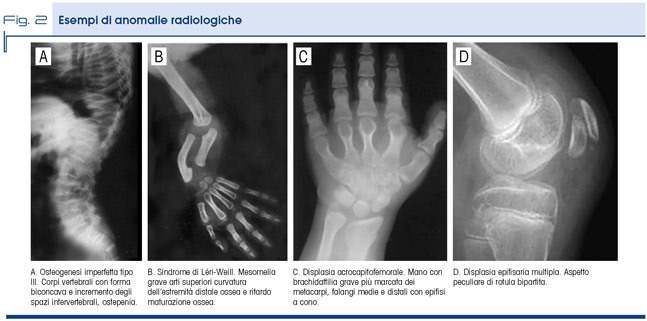
Altre condizioni
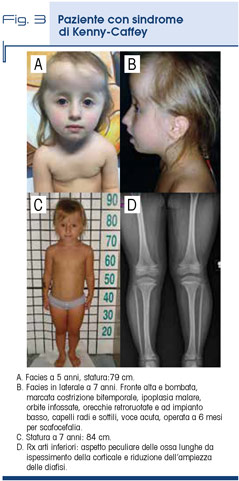
La sindrome di Kenny-Caffey è una condizione con trasmissione di tipo autosomico dominante ed è dovuta a mutazioni del gene FAM111A, che ha un ruolo nella replicazione del DNA ancora non ben precisato. Il fenotipo è caratterizzato da bassa statura grave ad esordio prenatale, craniostenosi, anomalie oculari (cataratta congenita, ipermetropia mar-cata, calcificazione retina), anemia, calcificazioni dei nuclei della base, epilessia, ipoparatiroidismo, ipocalcemia, scoliosi, brachidattilia mani e piedi. Facies con fronte bombata, ipertelorismo, costrizione bitemporale, ipoplasia malare, sovracciglia rade, naso piccolo, tip bulboso, labbro inferiore everso, voce con timbro acuto. La RX dello scheletro mostra anomalie diafisarie di tutte le ossa lunghe caratterizzate da ispessimento della corticale con riduzione di spessore della diafisi, più evidente agli arti inferiori rispetto ai superiori. Allelica alla tipo 2 è la osteocraniostenosi (nota anche come Gracile bone dysplasia) ma molto più grave (Tab. 3 e Fig. 3).


 Lo Spettro associato al gene SHOX. Si tratta di una serie di condizioni con bassa statura associate a aploinsufficienza del gene SHOX, causata da delezioni, mutazioni puntiformi, duplicazioni parziali del gene o delezioni in eterozigosi di enhancer localizzati upstream o downstream al gene. SHOX è localizzato nella regione pseudoautosomica del braccio corto del cromosoma X e del cromosoma Y che nello sviluppo embrionale è espresso soprattutto a livello degli arti; codifica per un fattore di trascrizione che agisce come inibitore della proliferazione cellulare ed è coinvolto nella regolazione di diversi pathway implicati nei meccanismi della GP.
Lo Spettro associato al gene SHOX. Si tratta di una serie di condizioni con bassa statura associate a aploinsufficienza del gene SHOX, causata da delezioni, mutazioni puntiformi, duplicazioni parziali del gene o delezioni in eterozigosi di enhancer localizzati upstream o downstream al gene. SHOX è localizzato nella regione pseudoautosomica del braccio corto del cromosoma X e del cromosoma Y che nello sviluppo embrionale è espresso soprattutto a livello degli arti; codifica per un fattore di trascrizione che agisce come inibitore della proliferazione cellulare ed è coinvolto nella regolazione di diversi pathway implicati nei meccanismi della GP.
Le manifestazioni cliniche legate all’aploinsufficienza di SHOX comprendono uno spettro che va da quadri più severi (discondrosteosi di Léri-Weill) alla bassa statura non sindromica. La Léri-Weill è caratterizzata dalla triade bassa statura, mesomelia degli arti e deformità di Madelung (curvatura dell’avambraccio per disallineamento di radio, ulna e ossa carpali a livello del polso). La bassa statura isolata senza anomalia di Madelung presenta un’ampia variabilità clinica sia inter- che intra-familiare. L’aploinsufficienza del gene SHOX è causa della bassa statura nelle pazienti con sindrome di Turner. Il riscontro di una variante patogenetica in SHOX è una delle indicazioni ad effettuare terapia con GH, molto efficace, senza necessità di praticare test da stimolo per identificare il deficit dell’ormone. Varianti bialleliche di SHOX sono causa della displasia mesomelica di Langer, caratterizzata da bassa statura grave con brevità marcata delle ossa lunghe, ipoplasia o aplasia di ulna e fibula e incurvamento di radio e tibia; non è presente la deformità di Madelung (Tab. 3).
Conclusioni
Il primo step, indispensabile, del percorso diagnostico è clinico-radiologico: la valutazione accurata dei segni clinici e radiologici può consentire almeno un inquadramento in uno dei 42 gruppi o famiglie e eventuali indagini genetiche mirate.
L’eterogeneità clinica e allelica di diverse condizioni in relazione a mutazioni in domain diversi di uno stesso gene e soprattutto in malattie estremamente rare possono complicare l’iter diagnostico. In questi casi è indispensabile ricorrere ad un’indagine molecolare mediante WES che consente di definire la diagnosi nella quasi totalità dei casi (92%).
È opportuno sottolineare l’importanza di estendere l’analisi mediante WES anche a casi di bassa statura idiopatica come dimostrato, ad esempio, dall’aver identificato mutazioni dominanti del gene NPR2 o del gene ACAN (Tab. 1,2) in casi del genere. In un recente articolo viene riportato che il percorso diagnostico limitato a tecniche di laboratorio senza una prioritaria valutazione clinico-radiologica consente di accertare la causa in circa il 17% dei casi che raggiunge il 33% con l’ausilio del WES.
Il livello di conoscenze attuali ha reso possibile poter diagnosticare con diversi criteri clinici, radiologici e molecolari la maggior parte delle attuali note 437 displasie scheletriche. Nello stesso tempo sono molto migliorate le condizioni di vita dei pazienti grazie ai progressi in campo terapeutico sia chirurgico, per prevenire le deformità, sia farmacologico con il ricorso a farmaci sia nuovi che già in uso per altre indicazioni sia con la terapia enzimatica sostitutiva (ad es. in alcune mucopolisaccaridosi) ma soprattutto con terapie specifiche (i trials già conclusi o in itinere nei casi di acondroplasia) o di recente la disponibilità di un anticorpo monoclonale, il burosumab, per i pazienti con la forma di rachitismo ipofosfatemico X-linked.
Bibliografia
Bibliografia
Review generali
- Wit JM, Ranke MB, Kelnar CJH. ESPE classification of Paediatric Endocrine diagnosis Short Stature. Horm Res 2007; 68 (sup.2):1-6.
- Scarano G. L’approccio alle Displasie Scheletriche: istruzioni per l’uso. Focus sulle Malattie Rare non diagnosticate. Il Pediatra 2020, febbraio. 46-51.
- Mortier GR, Cohn DH, Cormier-Daire V et al. Nosology and Classification of Genetic Skeletal Disorders: 2019 Revision. Am J Med Genet 2019; 179A:2393- 2419.
- Duarte SP, Rocha ME, Bidondo MP, Liascovich R et al. Bone dysplasias in 1.6 million births in Argentina. Eur J Med Genet 2019, 62 (12):103603.
- Barbosa-Buck CO, Orioli IM, da Graça Dutra M et al. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet 2012, 158A:1038-1045.
- Baldridge D, Shchelochkov O, Kelley B and Lee B. Signaling Pathways in Human Skeletal Dysplasias. Annu Rev Genomics Hum Genet 2010, 11:9.1–9.29
- Tsang KY, Tsang SW, Chan D, Cheah KSE. The Chondrocytic Journey in Endochondral Bone Growth and Skeletal Dysplasia. Birth Def Res (Part C) 2014, 102:52–73.
- Geister KA and Camper SA. Advances in Skeletal Dysplasia Genetics. Annu Rev Genomic Hum Genet 2015, 16:199-227
Articoli specifici
- Ornitz DM and Marie PJ. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev 2015; 29:1463-86.
- Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis. 2019;14(1):1.
- Salazar VS, Gamer LW, Rosen V. BMP Signalling in Skeletal Development, Disease and Repair. Nat Rev Endocrinol 2016; 12:203-21.
- Bacino CA. ROR2-Related Robinow Syndrome. In: Adam MP, Ardinger HH, Pagon RA et al. ed.s GeneReviews Seattle (WA): Univ. of Washington, 1993-2019.
- Roifman M, Brunner H, Lohr J et al. Autosomal Dominant Robinow Syndrome. In: Adam MP, Ardinger HH, Pagon RA et al. ed.s GeneReviews. Seattle (WA):Univ of Washington, 1993-2019.
- Schipani E, Langman CB, Parfitt AM et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. New Eng J Med 1996, 335:708 -714.
- Bartels CF, Bükülmez H, Padayatti P et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet 2004; 75:27-34.
- Miura K, Kim OH, Lee HR et al. Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am J Med Genet 2014;164A:156-63.
- Thompson SW, Merriman B, Funari VA et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C type lectin domain of aggrecan. Am J Hum Genet 2009; 84: 72-79.
- Gibson BG and Briggs MD. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. OJRD 2016; 11:86-93.
- Van Dijk FS, Sillence Do. Osteogenesis Imperfecta: Clinical Diagnosis, Nomenclature and Severity Assessment. Am J Med Genet 2014, 164A:1470-81.
- Clewemar P, Hailer NP, Hailer Y et al. Expanding the phenotypic spectrum of osteogenesis imperfecta type V including heterotopic ossification of muscle origins and attachments. Mol Genet Genomic Med 2019;7:e723.
- Gregersen PA, Savarirayan R, Adam MP et al. Ed.s. Type II Collagen Disorders Overview. In: GeneReviews. Seattle (WA): Univer. of Washington, Seattle; 1993–2020. 2019 Apr 25.
- Posey KL, Coustry F, Hecht JT. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol 2018, 71-72:161-173.
- Anthony S, Munk R, Skakun W, Masini M. Multiple Epiphyseal Dysplasia. J Am Acad Orthop Surg 2015; 23:164-172.
- Hanson-Kahn A, Li B, Cohn DH, Nickerson DA, Bamshad MJ, et al. Autosomal Recessive Stickler Syndrome Resulting From a COL9A3 Mutation. Am J Med Genet 2018; 176A:2887-2891.
- Unger S, Gorna MW, Le Bechec A et al. FAM111A Mutations Result in Hypoparathyroidism and Impaired Skeletal Development. Am J Hum Genet 2013; 92:990-5.
- Blaschke RJ, Rappold G. The Pseudoautosomal Regions, SHOX and Disease. Curr Opin Genet Dev 2006; 16:233-9.
- Bunyan DJ, Baffico M, Capone L et al. Duplications Upstream and Downstream of SHOX Identified as Novel Causes of Leri-Weill Dyschondrosteosis or Idiopathic Short Stature.Am J Med Genet 2016, 170A:949-57.
- Hauer NN, Popp B, Shoeller E et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet Med 2018;20:630-8.