




la Newsletter
Distrofia miotonica congenita: un esempio di presa in carico...
La distrofia miotonica congenita viene trasmessa prevalentemente...
Presentiamo qui il caso di una bambina con distrofia miotonica congenita (CMD) nata da madre affetta alla quale è stata posta diagnosi di distrofia miotonica di tipo 1 (DM1) (range di espansione E3) alla 29° settimana gestazionale presso il nostro Centro. Si tratta di una bambina di 2 anni e 10 mesi la cui madre è stata presa in carico presso il Centro clinico NeMO dalla sua diagnosi avvenuta al momento della gravidanza. Il parto è stato organizzato in team con i colleghi dei reparti di Ostetricia e Ginecologia e di Terapia Intensiva Neonatale (TIN) dell’Ospedale Niguarda, che ospita il nostro Centro. La bambina è nata da gravidanza insorta da FIVET normodecorsa fino all’ultimo mese, poi riscontro di polidramnios e riduzione dei MAF per cui è stato posto sospetto di DM1 anche nel feto. Il parto è avvenuto da TC a membrane integre alla 37+1 settimana (IA 5/7/9; P 3150 g, H48.5 cm, CC 38 cm).Alla nascita pianto iniziale, poi bradipnea e cianosi diffusa.
È stata ventilata mediante CPAP, successivamente trasferita in TIN in NCPAP con FiO2 30%, posizionato SOG per inefficace coordinazione suzione/deglutizione, scarsa gestione delle secrezioni e frequenti episodi di vomito.
A 5 gg di vita effettuati: ECG risultato nella norma, visita oculistica con riscontro di lieve ptosi palpebrale, rx del torace con riscontro di marcata ipoespansione polmonare e relaxatio diaframmatica bilaterale, eco addome con epatomegalia (>lobo sinistro); rene sinistro lievemente ptosico con lieve ipotonia delle cavità escretrici. A 7 gg di vita riscontro di colestasi trattata con acido ursodesossicolico.
A 9 gg di vita è stata effettuata indagine genetica per conferma della diagnosi. Eseguita consulenza NPI del Centro Nemo, segnalato pianto ipovalido, motricità spontanea estremamente povera, importante scialorrea, ipotono, postura a batrace. RT ipoevocabili simmetricamente.
La CMD è la forma più grave della DM1, ad esordio neonatale, i cui sintomi possono evidenziarsi anche prima della nascita. Viene trasmessa prevalentemente dalla madre affetta (spesso inconsapevole) al bambino, ma in una forma più grave (1). La DM1, è una malattia muscolare caratterizzata da miotonia e danni multiorgano, associati a debolezza muscolare di gravità variabile (2). La DM1 è la forma di distrofia muscolare più comune, con una prevalenza di 1/8.000 (3). Questa malattia si associa a mutazioni del gene DMPK sul cromosoma 19 che si associa ad una ripetizione anomala della tripletta CTG, fino a diverse migliaia (4). La trasmissione è autosomica dominante e può verificarsi il fenomeno dell’anticipazione (ossia la malattia tende ad aggravarsi e a manifestarsi più precocemente nelle generazioni successive) (5-6).
Il programma riabilitativo
È stata impostata presa in carico riabilitativa in TIN in collaborazione con il Centro Clinico NeMO: prevista Care posturale per favorire la funzionalità respiratoria e la motricità, promuovere il sonno, diminuire il reflusso gastro-esofageo.
A 36 gg di vita per garantire il corretto allineamento dei piedi, spontaneamente mantenuti in equino-varo-supinato, è stato eseguito confezionamento di tutori gamba-piede in vetroresina.
A 95 gg di vita la bambina è stata dimessa dalla TIN ed è stata accolta al Centro Clinico NeMO per una presa in carico di tipo multidisciplinare ed il proseguimento del programma riabilitativo.
 Eseguito adattamento alla NIV notturna e alla macchina della tosse. Sono stati confezionati nuovi tutori AFO di posizionamento in materiale termoplastico (polipropilene) su misura ed è stato prescritto sistema di postura per il mantenimento della posizione seduta, per assenza del controllo del tronco. A 115 gg di vita eseguito presso la U.O. Otorinolaringoiatria di Niguarda studio fibroendoscopico, con evidenza di una deglutizione non funzionale con penetrazione ed aspirazione silente.
Eseguito adattamento alla NIV notturna e alla macchina della tosse. Sono stati confezionati nuovi tutori AFO di posizionamento in materiale termoplastico (polipropilene) su misura ed è stato prescritto sistema di postura per il mantenimento della posizione seduta, per assenza del controllo del tronco. A 115 gg di vita eseguito presso la U.O. Otorinolaringoiatria di Niguarda studio fibroendoscopico, con evidenza di una deglutizione non funzionale con penetrazione ed aspirazione silente.
A 154 gg di vita eseguito intervento di plastica antireflusso secondo Nissen e posizionamento di PEG. È stata inoltre effettuata attività di coaching multidisciplinare per educare il caregiver alla gestione delle attività di vita quotidiana: igiene, nutrizione, proposta di attività ludiche e stimolazioni neurosensoriali, gestione delle ortesi/ausili, organizzazione della casa. Dimessa a 6 mesi di età, con indicazione ad utilizzo di NIV notturna e nutrizione enterale a boli ed attività riabilitativa (logopedia 1v/sett e fisioterapia 1v/sett).
Il follow-up
 Ai fini di un corretto monitoraggio della paziente è stato impostato regolare follow-up presso il Centro NeMO. A 13 mesi la bimba ha mostrato un buon controllo del capo con possibile mantenimento della posizione seduta in cifosi se stabilizzata al cingolo pelvico.
Ai fini di un corretto monitoraggio della paziente è stato impostato regolare follow-up presso il Centro NeMO. A 13 mesi la bimba ha mostrato un buon controllo del capo con possibile mantenimento della posizione seduta in cifosi se stabilizzata al cingolo pelvico.
Presenti vocalizzi ed accenno di lallazione. Nutrizione esclusiva via PEG.
A 20 mesi è stato realizzato un corsetto statico equilibrato per correggere l’ipercifosi e, dopo aver effettuato RX anche che denotava sfuggenza e sublussazione della testa femorale di sinistra, è stata prescritta statica da prona per utilizzo con corsetto e tutori AI.
A 21 mesi è stata raggiunta la posizione seduta autonoma ed è stata prescritta carrozzina manuale superleggera. La piccola ha iniziato l’assunzione di cucchiaini di omogenizzati sotto supervisione della logopedista (Fig. 1).
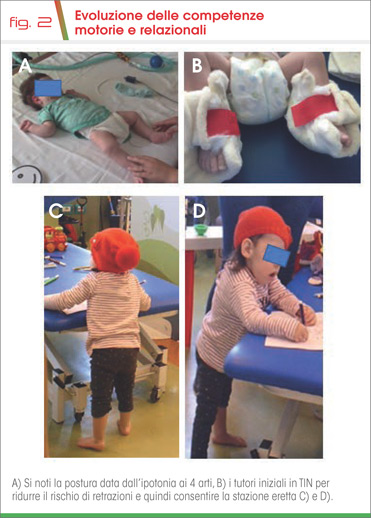
Attualmente, a 32 mesi, la bambina è in grado di pronunciare alcune parole correttamente, anche se permane importante ipotono ed ipostenia della muscolatura del volto. Le competenze motorie e relazionali appaiono invece in evoluzione positiva: la bambina effettua qualche passo con il deambulatore sotto la guida della terapista, migliorata la volontà comunicativa (Fig. 2).
Conclusioni
La collaborazione multidisciplinare fra i reparti di Ginecologia, Terapia Intensiva Neonatale dell’Ospedale Niguarda ed il Centro Clinico NeMo ha sostenuto la madre durante la gravidanza ed ha permesso la presa in carico precoce della bambina.
Bibliografia
- Bird TD. Myotonic Dystrophy Type 1. 1999 Sep 17 [updated 2021 Mar 25]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. PMID: 20301344.
- Soltanzadeh P. Myotonic Dystrophies: A Genetic Overview. Genes (Basel). 2022;13(2):367.
- Suominen T, Bachinski LL, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19(7):776-82.
- Harley HG, Rundle SA, MacMillan JC, et al. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet. 1993;52(6):1164-74.
- Ashizawa T, Dunne CJ, Dubel JR, et al. Anticipation in myotonic dystrophy. I. Statistical verification based on clinical and haplotype findings. Neurology. 1992;42(10):1871-7.
- Meola G, Cardani R. Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. J Neuromuscul Dis. 2015;2(s2):S59-S71.