




la Newsletter

Anna Capasso, Gianpaolo Cicala, Martina Ricci, Eugenio Mercuri
Neuropsichiatria Infantile, Policlinico Gemelli IRCCS, Università Cattolica, Roma
Distrofia muscolare di Duchenne: nuovi standard di cura e...
CK sieriche e/o transaminasi elevate sono importanti per...
La distrofia muscolare di Duchenne (DMD) è la più frequente distrofia muscolare in età pediatrica, con un’incidenza annuale di 1/5000 maschi nati vivi ed una prevalenza stimata di 8-29/100000 maschi. (1) Circa il 60–70% di mutazioni nel gene DMD sono delezioni, il 5–15% duplicazioni, e il 20% sono piccole mutazioni.
I ragazzi affetti da DMD tipicamente sviluppano i sintomi della malattia dopo il primo anno di vita, spesso in seguito ad un lieve ritardo nell’acquisizione della deambulazione. I primi sintomi sono rappresentati da cadute frequenti, incapacità di correre, salire le scale e difficoltà nell’alzarsi da terra, con necessità di fare forza con le braccia poggiandole sulle ginocchia (manovra di Gowers). È comune, inoltre, un ritardo nell’acquisizione del linguaggio (50%) e nel 30% dei pazienti sono presenti comorbilità, quali disabilità intellettiva, disturbo di spettro autistico e deficit di attenzione.
Criteri diagnostici
Una diagnosi di DMD dovrebbe essere sospettata in bambini, soprattutto prima dei 5 anni, che hanno un ritardo psicomotorio, debolezza, ipertrofia dei polpacci e difficoltà ad alzarsi da terra senza appoggiarsi (segno di Gowers). Altri segni di ritardo, come ritardo del linguaggio sono anche frequenti. L’esame delle CK sieriche è spesso dirimente e spesso la diagnosi viene effettuata per riscontro casuale di elevate CK e/o transaminasi, osservate durante prelievi eseguiti per altri motivi intercorrenti. Livelli elevati di CK sono importanti per sospettare la diagnosi di DMD ma non sono specifici ed è importante che la diagnosi venga sempre effettuata tramite test genetico. Il test più comunemente usato è MLPA che consente di identificare delezioni e duplicazioni presenti in oltre il 70 % dei pazienti. Se MLPA, o array CGH che viene usato sempre più frequentemente, non identificano la mutazione, è necessario completare l’esame usando Sanger sequencing con sequenziamento dei 79 esoni e delle regioni introniche alla ricerca di mutazioni puntiformi o altre piccole mutazioni. La biopsia muscolare, in passato esame principe per la diagnosi di DMD, è ormai per lo più indicata nei casi di fenotipi discordanti con la mutazione o di mutazioni compatibili con fenotipi variabili.
Il coinvolgimento distrofico del muscolo resta relativamente stabile fino ai 7 anni circa, quando la progressione diventa più rapida. Classicamente la perdita della deambulazione autonoma avviene entro i 12 anni (media di 9.5 anni); in seguito possono insorgere scoliosi, progressiva perdita della funzionalità degli arti superiori, insufficienza respiratoria e cardiomiopatia dilatativa. Queste complicanze portano ad un’età media di sopravvivenza che si attesta intorno alla tarda adolescenza.
Con la recente revisione degli standard di cura, e soprattutto il largo utilizzo dei corticosteroidi (2) attualmente la deambulazione è conservata fino all’età media di 13-14 anni. Studi recenti suggeriscono che pazienti trattati con un regime quotidiano di corticosteroidi hanno una perdita della deambulazione più tardiva rispetto a quelli con regime intermittente (3).
Parallelamente grazie alla revisione degli standard di cura con l’implementazione del trattamento multidisciplinare e in particolare degli aspetti cardiologici e respiratori, l’età media di sopravvivenza si è spostata alla fine della seconda decade di vita. Lo scompenso cardiaco dovuto alla progressiva cardiomiopatia dilatativa è, al momento, la principale causa di morte. Da anni sono stati utilizzati in maniera profilattica inibitori dell’enzima di conversione dell’angiotensina (ACE-inbitori) e/o beta-bloccanti per il trattamento precoce della cardiomiopatia dilatativa nei pazienti DMD, generalmente dopo i 10 anni di età o alla comparsa dei primi segni di ridotta funzionalità. (4) Non vi è ancora un consenso sull’utilizzo di trattamenti farmacologici ai fini di profilassi cardiaca in pazienti di età inferiore ai 10 anni; tale approccio è al momento supportato da studi condotti su coorti ristrette, (5) mentre sono in corso studi randomizzati su popolazioni più estese (EudraCT 2007-005932-10). Recenti studi riportano benefici dall’utilizzo aggiuntivo dell’eplerenone, un anti-mineralcorticoide della famiglia dello spironolattone.
Parallelamente a questi aspetti, sono state anche implementate nuove raccomandazioni su diversi aspetti con particolare attenzione agli aspetti endocrinologici e nutrizionali ed alla gestione dei fatti acuti, sottolineando l’importanza di una medicina anticipatoria.
Nelle ultime due decadi particolare attenzione è stata dedicata al coinvolgimento cerebrale che, al contrario della degenerazione dei muscoli scheletrici e del cuore, non sembra essere progressivo nella DMD. Circa un terzo dei pazienti presenta disabilità intellettiva e il quoziente intellettivo nei ragazzi DMD è in media una deviazione standard al di sotto dei dati normativi età specifici. Negli ultimi anni si è prestata particolare attenzione alla comorbidità con altri aspetti di interessamento del SNC con evidenza di altri disordini del neurosviluppo come ADHD e disturbo dello spettro autistico ed una alta percentuale di disturbi psichiatrici quali ansia e altri disturbi internalizzanti.
L’interessamento cerebrale può essere parzialmente ascrivibile alla presenza di diverse isoforme della distrofina in tale sede, la cui diversa espressione è determinata dal sito della mutazione del gene DMD.
Con l’avvento dei trial clinici si è resa manifesta la necessità di validazione delle misure di outcome e di dati provenienti da studi longitudinali di storia naturale. Il test del cammino dei sei minuti (6MWT) è stato utilizzato come misura primaria di outcome in molti trial clinici; inoltre, sono state largamente utilizzate anche scale funzionali malattia-specifiche, quali la North Star Ambulatory Assessment (NSAA) o la Performance of Upper Limb (PUL). Studi di storia naturale hanno mostrato un’ampia variabilità nella progressione della DMD, anche entro 12 mesi, e che l’andamento di malattia non è lineare (6,7). I differenti pattern di progressione di malattia possono essere identificati grazie alla combinazione di valutazioni funzionali (es. 6MWT con test funzionali a tempo) e di imaging (Risonanza Magnetica).
Approcci terapeutici sperimentali
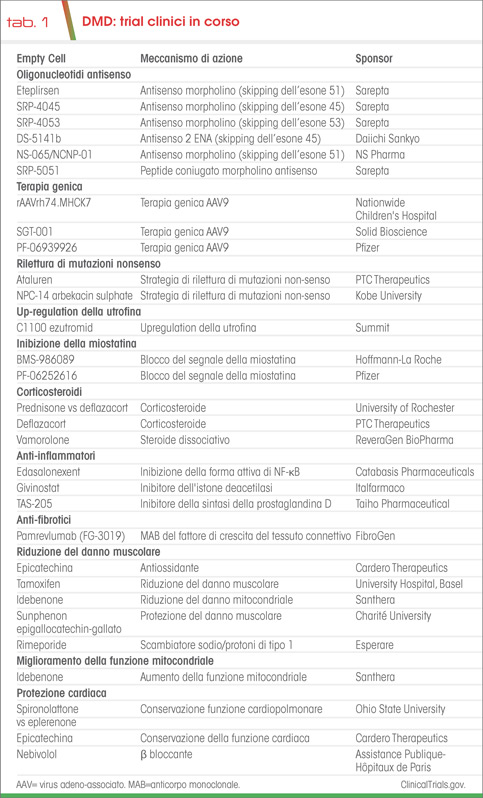
Dal punto di vista terapeutico, la scorsa decade ha visto un aumento esponenziale degli approcci sperimentali per la DMD, con l’approvazione di nuovi farmaci sia negli Stati Uniti che in Europa. Attualmente ci sono più di 20 trial clinici in corso per la distrofia muscolare di Duchenne, dalla fase 1 a studi più avanzati.
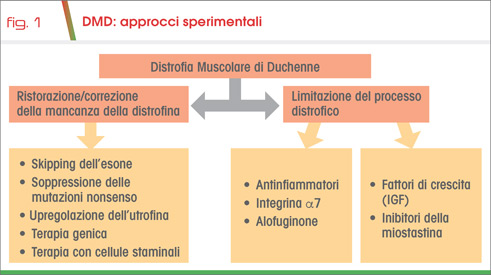 Gli approcci utilizzati si basano sul miglioramento dell’integrità strutturale delle fibre muscolari o sul ripristino della produzione di distrofina, oppure sono mirati al trattamento delle conseguenze della carenza di distrofina – ad esempio i processi infiammatori, la fibrosi, la rigenerazione muscolare e la massa muscolare (Fig.1).
Gli approcci utilizzati si basano sul miglioramento dell’integrità strutturale delle fibre muscolari o sul ripristino della produzione di distrofina, oppure sono mirati al trattamento delle conseguenze della carenza di distrofina – ad esempio i processi infiammatori, la fibrosi, la rigenerazione muscolare e la massa muscolare (Fig.1).
Nell’ambito delle strategie utilizzate per ripristinare l'espressione della distrofina nel sarcolemma, si annovera l’utilizzo di piccole molecole o oligonucleotidi antisenso, ed approcci di terapia genica con virus adenoassociati (AAV).
Un altro approccio terapeutico è indirizzato alle mutazioni nonsenso. Ataluren, una piccola molecola assunta per via orale, consente al ribosoma la rilettura di mutazioni nonsenso e ripristina parzialmente la produzione di distrofina. Due studi randomizzati, in doppio-cieco, placebo-controllo hanno mostrato un trend di efficacia terapeutica misurata attraverso il 6MWT, sebbene entrambi gli studi non abbiano raggiunto l’endpoint primario delle 48 settimane. Il disegno dello studio potrebbe avere avuto un ruolo in tale risultato, dato che entrambi gli studi hanno reclutato un alto numero di ragazzi con DMD in una fase di stabilità di malattia, che potrebbe aver ridotto la possibilità di apprezzare l’efficacia del trattamento. Infatti, quando si considerano solo i ragazzi che stanno perdendo la deambulazione (endpoint specifico degli studi di fase 3), si è documentata una differenza statisticamente significativa tra il gruppo dei bambini trattati e il gruppo placebo (8). Sulla base di questi risultati e del buon profilo di tollerabilità, l’Agenzia Europea del Farmaco (EMA) ha garantito un’approvazione condizionale in attesa che vengano condotti ulteriori trial.
Una strategia di trattamento alternativa per le delezioni out-of-frame è rappresentata dagli oligonucleotidi antisenso. Tali molecole devono essere somministrate sistematicamente ogni settimana, per consentire la manipolazione del processo di splicing del pre-mRNA nei pazienti eleggibili e per generare un messaggio in-frame rimuovendo un esone adiacente alla delezione breakpoint (es. frame restoring exon skipping). Questo approccio è mutazione specifico e facilitato dalla clusterizzazione delle delezioni in hotspot specifici: lo skipping dell’esone 51, per esempio, è applicabile a circa il 13% di tutti i ragazzi con delezioni. Nei trial della DMD sono stati utilizzati due tipi differenti di varianti chimiche di oligonucleotidi antisenso, il morpholino e il 2’OMe. Entrambi determinano il ripristino della produzione di distrofina quando somministrati in singola iniezione intramuscolo nei ragazzi con DMD. In due successivi studi di fase 2, l’oligonucleotide antisenso 2’OMe, drisapersen, ha raggiunto l’endpoint clinico. Tuttavia, in un più ampio studio di fase 3 non ha mostrato un beneficio clinico. I criteri di inclusione potrebbero aver avuto un ruolo; una recente analisi post-hoc ha suggerito un miglioramento nel 6MWT in favore di drisapersen nei bambini con declino moderato, rispetto ai pazienti che ricevevano placebo. Nondimeno, la scarsa efficacia clinica unita ad un profilo di tollerabilità sfavorevole ha portato ad abbandonare lo sviluppo del 2’OMe per la DMD.
In uno studio di fase 2, l’oligonucleotide antisenso morpholino disegnato per lo skipping dell’esone 51 (eteplirsen), induce il ripristino della distrofina quando somministrato in infusione endovenosa. Studi successivi hanno confermato la produzione di distrofina in molti dei pazienti trattati, e una differenza basata sul 6MWT nel decorso clinico tra i bambini trattati e gli studi di storia naturale. (9) Questi dati hanno portato la Food and Drug Administration (FDA) ad approvare eteplirsen in maniera condizionata negli Stati Uniti nonostante le criticità dovute alla ristretta popolazione di pazienti trattati, la modesta quantità di distrofina prodotta, (9) e l’assenza di un gruppo placebo. Recentemente sono stati pubblicati ulteriori dati a lungo termine (5 anni) sulla preservazione della funzione respiratoria nei ragazzi con DMD. Attualmente sono in corso altri studi che hanno come bersaglio l’esone 51, 45 ed il 53.
Inoltre, sono in corso studi di fase 1 sugli oligonucleotidi antisenso di nuova generazione: un peptide coniugato oligonucleotide antisenso morpholino per migliorare la specificità per il tessuto muscolare; una nuova variante stereochimica del 2’OMe, che dovrebbe determinarne una maggiore efficacia, consentendone un minor dosaggio e minori eventi avversi.
 La terapia genica con vettore adenovirale (AAV) è un’altra opzione terapeutica per la distrofia muscolare di Duchenne. La grandezza del gene DMD non consente di includere l’intero cDNA nei vettori AAV. Comunque, sono state sviluppate versioni considerevolmente ridotte di microdistrofina che possono essere accomodate nei vettori AAV. Dal momento che l’arrivo attraverso il vettore virale al muscolo è associato ad una risposta immunitaria verso l’AAV che preclude la possibilità di ripetere la somministrazione con lo stesso sierotipo, è essenziale sviluppare strategie efficienti e con una prospettiva realistica di produrre un beneficio terapeutico per i pazienti, al fine di ridurre la necessità di ripetere il trattamento. Recenti studi preclinici hanno dimostrato il potenziale di AAV8 e AAV9 di raggiungere il tessuto muscolare, con un miglioramento dal punto di vista sia istologico, che clinico (10). Molti gruppi accademici ed industriali hanno intrapreso trial clinici di fase 1 in cui dosi crescenti di AAV9 e AAVhr74 vengono somministrati sistematicamente in ragazzi con DMD.
La terapia genica con vettore adenovirale (AAV) è un’altra opzione terapeutica per la distrofia muscolare di Duchenne. La grandezza del gene DMD non consente di includere l’intero cDNA nei vettori AAV. Comunque, sono state sviluppate versioni considerevolmente ridotte di microdistrofina che possono essere accomodate nei vettori AAV. Dal momento che l’arrivo attraverso il vettore virale al muscolo è associato ad una risposta immunitaria verso l’AAV che preclude la possibilità di ripetere la somministrazione con lo stesso sierotipo, è essenziale sviluppare strategie efficienti e con una prospettiva realistica di produrre un beneficio terapeutico per i pazienti, al fine di ridurre la necessità di ripetere il trattamento. Recenti studi preclinici hanno dimostrato il potenziale di AAV8 e AAV9 di raggiungere il tessuto muscolare, con un miglioramento dal punto di vista sia istologico, che clinico (10). Molti gruppi accademici ed industriali hanno intrapreso trial clinici di fase 1 in cui dosi crescenti di AAV9 e AAVhr74 vengono somministrati sistematicamente in ragazzi con DMD.
Numerosi trial clinici di fase 1-3 sono in corso per valutare la tollerabilità e l’efficacia di:
- targeting specifico della via infiammatoria del NF-kB, che mira a mantenere l’efficacia corticosteroidea, riducendone gli eventi avversi cronici;
- down-regolation della via della miostatina, che nei modelli animali induce un’aumentata rigenerazione ed aumento della massa muscolare;
- composti con un effetto sulla fibrosi e la rigenerazione muscolare;
- incrementi delle risorse bioenergetiche mitocondriali (Tab. 1).
L’insieme dei nuovi standard di cura e le nuove prospettive terapeutiche rendono il campo della distrofia muscolare di Duchenne estremamente interessante. Nonostante nessuno dei tentativi terapeutici e di standard di cura finora disponibile sia riuscito a sovvertire completamente l’andamento progressivo della malattia, vi è evidenza di come diverse componenti, dall’introduzione delle modalità di ventilazione non invasiva alla profilassi cardiologica, dall’uso dei corticosteroidi ai monitoraggi nutrizionali, abbiano significativamente allungato la sopravvivenza e posticipato o in alcuni casi annullato la comparsa di complicanze della malattia.
Un esempio classico è legato alla scoliosi. In passato, quando i bambini perdevano la deambulazione autonoma ad un’età media di 8-9 anni, tutti, invariabilmente, trovandosi in carrozzina al momento dello scatto puberale, sviluppavano una scoliosi grave che veniva sempre trattata chirurgicamente. Il posticiparsi dell’età in cui viene persa la deambulazione autonoma ha ridotto notevolmente il rischio di una scoliosi grave e al momento la chirurgia vertebrale viene eseguita in pochissimi casi. L’arrivo della ventilazione non invasiva ha fatto ridurre drasticamente il numero di morti per cause respiratorie nella seconda decade ed al momento la causa più frequente di mortalità è legata alla componente cardiaca, nella decade successiva.
L’arrivo di nuovi farmaci come ataluren, disponibile in Italia ed in Europa, sembra determinare un ulteriore rallentamento della progressione della malattia. I dati preliminari delle nuove terapie, come la terapia genica, sembrano indicare un ulteriore trend di stabilità che, se confermato dal follow-up a lungo temine, potrebbe significare una nuova modifica della storia naturale della malattia.
Bibliografia
- Norwood FL, Harling C, Chinnery PF,et al. Prevalence of genetic muscle disease in northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132(Pt11):3175-86.
- Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267.
- Guglieri M, Bushby K, McDermott MP, et al. Effect of Different Corticosteroid Dosing Regimens on Clinical Outcomes in Boys With Duchenne Muscular Dystrophy: A Randomized Clinical Trial. JAMA. 2022;327(15):1456-1468.
- Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(2):153–61.
- Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154(3):596–602.
- Henricson E, Abresch R, Han JJ, et al. The 6-Minute Walk Test and person-reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013;5:ecurrents.md.9e17658b007eb79fcd6f723089f79e06..
- Pane M, Mazzone ES, Sivo S, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One. 2014;9(10):e108205.
- Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–87.
- Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2): 257–71.
- Chamberlain JR, Chamberlain JS. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol Ther. 2017;25(5):1125–31.