




la Newsletter
Epilessia correlata a PCDH19 e sindrome di Dravet: fenotipi...
Il gene PCDH19, dopo il gene SCN1A, è il secondo gene più...
Descriviamo il caso clinico di una bambina arrivata alla nostra osservazione per la comparsa di crisi epilettiche ricorrenti, configuranti uno stato epilettico (SE). La bambina, primogenita da genitori non consanguinei, era nata a termine dopo una gravidanza normale senza presentare problemi perinatali. All’anamnesi risultava una familiarità positiva per ritardo di linguaggio, disturbi psichiatrici ed epilessia in età infantile non meglio specificata.
A 8 mesi la bambina, in condizioni di apparente benessere clinico, presentò degli episodi critici ricorrenti in veglia con arresto del contatto, sguardo fisso con espressione di paura, deviazione dello sguardo a lato alterno, cianosi periorale, ipertono talora seguito da movimenti clonici agli arti superiori. Le crisi duravano 2-3 minuti, più volte al giorno, configurando uno SE.
Al momento del ricovero, la bambina presentava un’ipotonia associata a ritardo psicomotorio globale, con maggiore compromissione nell’area comunicativa e linguistica. L’elettroencefalogramma (EEG) documentava un’attività di fondo lenta diffusa in banda theta-delta che manteneva una maggiore organizzazione nel sonno; presenti rare anomalie parossistiche focali prevalenti all’emisfero sinistro.
Gli esami biochimici (indici infiammatori, analisi del liquor, indagini metaboliche), di imaging cerebrale e genetici (cariotipo, Array CGH), risultarono normali. In considerazione del quadro di epilessia a esordio precoce e ritardo psicomotorio si decise di avviare un’indagine genetica, mediante Nex Generation Sequencing (NGS), su un pannello di geni coinvolti nelle forme di encefalopatia epilettica a esordio precoce.
Venne iniziata terapia antiepilettica d’attacco con utilizzo consecutivo di diazepam, fenobarbital e dintoina, con parziale risposta clinica (riduzione della frequenza delle crisi).
L’EEG in trattamento mostrava una discreta organizzazione dell’attività elettrica sia in veglia che in sonno, associata a un’attività epilettiforme focale sulle derivazioni centrali di sinistra con registrazione di crisi elettrocliniche ripetute a partenza focale centro-temporale sinistra (Fig. 1).
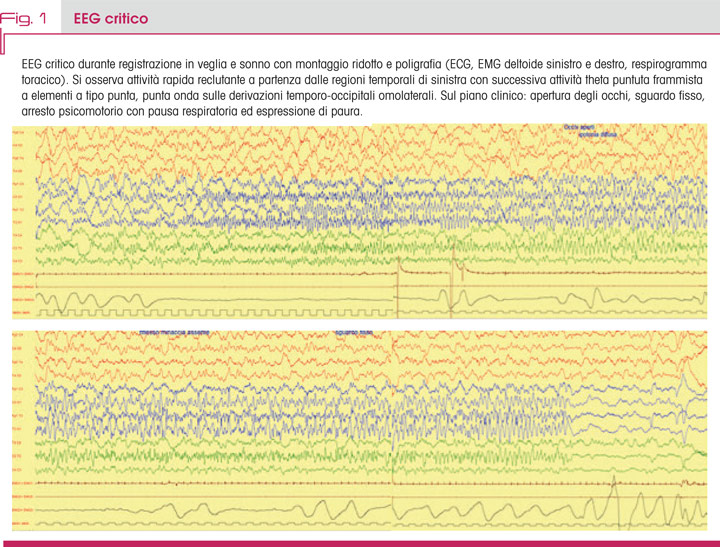
Fu poi iniziata una terapia di mantenimento per via orale prima con carbamazepina e poi con topiramato, ottenendo un controllo delle crisi.
All’età di 10 mesi la bambina presentò nuovi episodi critici in corso di febbre, semeiologicamente sovrapponibile ai precedenti. All’EEG non erano evidenti sostanziali modificazioni. La terapia d’attacco con fenitoina ev fu efficace nel controllo della crisi.
Successivamente, all’età di 12, 13, 14 e 15 mesi, la bambina presentò nuovi episodi critici in apiressia con ricorrenza in cluster pluriquotidiani. L’EEG documentava una discreta organizzazione dell’attività elettrica, associata alla presenza di anomalie epilettiformi a espressione focale prevalenti a sinistra (Fig. 2).
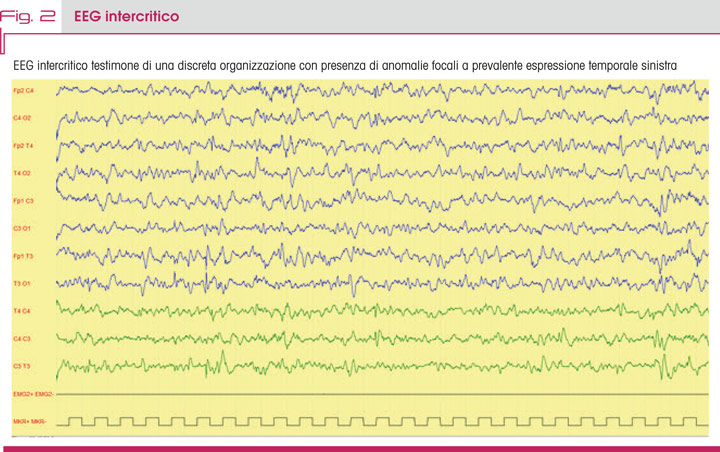
L’analisi molecolare, infine, documentò la presenza di una variante patogenetica di tipo troncante (p.Tyr366Leufs10) del gene PCDH19 (NM_00184880:C1091dupC). Il gene, localizzato sul cromosoma x (locus Xp22.1), codifica per la proteina Protocaderina.
Il trattamento sintomatico per os con clobazam in associazione a topiramato e dintoina, si dimostrò efficace; la dintoina fu sospesa dopo completo controllo delle crisi.
Attualmente la bambina ha 25 mesi e risulta libera da crisi. La valutazione neuropsichiatrica documenta un’evoluzione positiva delle competenze psicomotorie, in particolare nell’ambito dell’area comunicativo-sociale e motoria.
Sindrome da mutazione del gene PCDH19
La sindrome da mutazione del gene PCDH19 è stata descritta per la prima volta nel 2008 (1) in pazienti di sesso femminile con epilessia associata a ritardo cognitivo e disturbi comportamentali di grado variabile (EFMR, Female-restricted epilepsy and mental retardation) [OMIM 300088]. La mutazione PCDH19 è trasmessa da soggetti di sesso maschile portatori asintomatici, suggerendo una modalità di trasmissione X-linked "inusuale". Il meccanismo patogenetico, ad oggi, rimane sconosciuto. In letteratura sono state proposte diverse ipotesi (presenza di un gene omologo sul cromosoma Y con ruolo protettivo o "interazione" tra diverse popolazioni cellulari che esprimono le due varianti, mutata e normale, del gene) (2). La mutazione può insorgere con modalità de novo. In entrambi i casi (familiari o sporadici) la penetranza risulta incompleta, rendendo conto dell’ampia variabilità nell’espressione del fenotipo clinico. Il gene PCDH19 codifica per la proteina Protocaderina 19 coinvolta nei processi di adesione, migrazione cellulare e formazione delle connessioni sinaptiche nel corso dei processi di maturazione cerebrale (3).
Le crisi epilettiche hanno un esordio nel primo anno di vita, si presentano con frequenza elevata e mostrano farmacoresistenza. Lo sviluppo psicomotorio usualmente normale all’esordio, presenta una regressione successiva alla comparsa delle crisi con disabilità cognitiva di grado variabile, spesso medio-severo (70% dei casi). Si associano disturbi sul piano comportamentale e psichiatrico (disturbi dello spettro autistico nel 25% dei casi, disturbi ossessivi compulsivi, disturbi di ansia), evidenti e significativi anche dopo la scomparsa delle manifestazioni critiche che tendono a scomparire verso i 10-12 anni (4, 5).
In seguito alla diffusione delle nuove tecniche di indagine genetica, mutazioni a carico del gene PCDH19 sono state identificate in alcuni pazienti con sindrome di Dravet risultati negativi alla ricerca della mutazione genetica SCN1A (6).
Sindrome di Dravet
La sindrome di Dravet (DS) è una encefalopatia caratterizzata dalla comparsa di crisi epilettiche nel primo anno di vita favorite da stati febbrili. Le crisi sono cloniche/toniche-cloniche, monolaterali e generalizzate. È comune uno SE generalizzato o emiclonico. Altri tipi di crisi (di solito mioclonie, assenze atipiche, crisi parziali complesse) si manifestano a partire dall’età di 2-3 anni. Nel corso della malattia la durata delle crisi tende a ridursi a fronte di una frequenza aumentata, con sviluppo di farmacoresistenza nella maggioranza dei pazienti. Fattori scatenanti sono la chiusura degli occhi o la stimolazione con luce intermittente. Durante i primi stadi della malattia, l'EEG è normale, le anomalie (onde appuntite e con più punte generalizzate) compaiono a partire dai 2-3 anni. Il decorso è caratterizzato da ritardo dello sviluppo psicomotorio e dalla comparsa di disturbi del comportamento e atassia.
.jpg) Lo spettro clinico associato a mutazione PCDH19 si sovrappone a quello della DS. Le due sindromi epilettiche condividono alcune caratteristiche comuni ma allo stesso tempo mostrano segni clinici peculiari, che risultano utili nella diagnosi differenziale delle due forme (7) (Tab. 1).
Lo spettro clinico associato a mutazione PCDH19 si sovrappone a quello della DS. Le due sindromi epilettiche condividono alcune caratteristiche comuni ma allo stesso tempo mostrano segni clinici peculiari, che risultano utili nella diagnosi differenziale delle due forme (7) (Tab. 1).
Caratteristiche cliniche comuni: l’età di esordio dell’epilessia nel primo anno di vita, la presenza di uno sviluppo psicomotorio normale prima delle crisi, la sensibilità alla febbre quale fattore scatenante, la progressiva comparsa di un ritardo cognitivo e comportamentale.
Caratteristiche differenti: l’incidenza nei due sessi (il fenotipo EFMR è prevalente nelle femmine, a differenza del fenotipo DS in cui entrambi i sessi sono interessati), l’età di esordio delle prime crisi (nella EFMR verso i 6-36 mesi, con uno spettro più ampio rispetto al fenotipo Dravet-like, dove solitamente la prima crisi avviene tra i 5 e gli 8 mesi), il tipo di crisi prevalenti all’esordio e la modalità di espressione ed alcuni elementi elettroencefalografici. Riguardo il tipo di crisi, viene riportata una più alta incidenza di crisi in cluster e di breve durata nel fenotipo EFMR rispetto alla forma Dravet-like, che presenta generalmente crisi più lunghe e configuranti uno SE.
Crisi ipomotorie con componente affettiva (iniziale espressione di paura) sono tipiche della EFMR rispetto alla DS, in cui le crisi all’esordio sono prevalentemente di tipo clonico o emiclonico, con successiva predominanza di crisi miocloniche (8).
La storia naturale mostra un’evoluzione in genere positiva nella forma EFMR, con riduzione o regressione delle crisi dopo alcuni anni rispetto alla forma DS, in cui le crisi persistono con frequenza elevata e può svilupparsi farmacoresistenza.
Riguardo le caratteristiche EEG, è presente una più alta incidenza di anomalie epilettiformi nella forma DS, prevalentemente localizzate nelle regioni posteriori, rispetto alla forma EFMR, dove l’area prevalente è quella temporale. La fotosensibilità è specifica del fenotipo DS.
Il trattamento farmacologico nella sindrome EFMR risulta difficile soprattutto nei primi anni di vita, quando le crisi tendono a comparire con alta frequenza e risultano resistenti a molteplici terapie farmacologiche. Allo stato attuale, in letteratura esistono dati limitati, sia in termini di casistiche che di durata del follow up, sul trattamento farmacologico in questa forma di epilessia e non vi è un consenso su specifiche raccomandazioni terapeutiche (9).
Un recente lavoro multicentrico europeo su un elevato numero di pazienti ha dimostrato l’efficacia clinica dei farmaci bromide e clobazam, con una risposta positiva mantenuta nel tempo (10). In particolare clobazam ha raggiunto il completo controllo delle crisi nel 40% dei casi e una riduzione delle stesse superiore al 50% nel 6% dei casi. Dopo 12 mesi di trattamento, il 47% dei pazienti ha mantenuto una risposta clinica con una riduzione delle crisi superiore al 50%. L’uso di clobazam come farmaco add-on a valproato di sodio o topiramato risulta efficace e viene raccomandato come indicazione terapeutica anche nella DS (11).
Conclusioni
Il gene PCDH19, dopo il gene SCN1A, è il secondo gene più frequentemente coinvolto nell’insorgenza di epilessia a esordio in età infantile precoce in soggetti di sesso femminile.
 Elementi clinici suggestivi in senso diagnostico sono: crisi epilettiche nel primo anno di vita, prevalentemente di tipo focale, scatenate dalla febbre, farmacoresistenti all’esordio, con ritardo cognitivo ed elevata incidenza di regressione, successiva all’esordio delle crisi, e disturbi comportamentali associati (in prevalenza disturbi dello spettro autistico).
Elementi clinici suggestivi in senso diagnostico sono: crisi epilettiche nel primo anno di vita, prevalentemente di tipo focale, scatenate dalla febbre, farmacoresistenti all’esordio, con ritardo cognitivo ed elevata incidenza di regressione, successiva all’esordio delle crisi, e disturbi comportamentali associati (in prevalenza disturbi dello spettro autistico).
Nei soggetti con fenotipo clinico suggestivo di sindrome di Dravet è utile procedere alla ricerca della mutazione di questo gene, essendo responsabile di circa il 25% dei casi risultati negativi alla mutazione per il gene SCN1A.
Il riconoscimento delle peculiari caratteristiche cliniche ed elettroencefalografiche rappresenta un criterio utile per una diagnosi precoce, la formulazione di un giudizio prognostico e la definizione di strategie terapeutiche mirate (Tab. 1).
Nonostante l’elevata incidenza di farmacoresistenza nei pazienti con mutazioni del gene PCDH19 (specie nella fase d’esordio delle crisi), alcuni farmaci come il clobazam e i bromidi risultano maggiormente efficaci.
Bibliografia
- Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40(6):776-81.
- Depienne C, LeGuern E. PCDH19 related Infantile Epileptic Encephalopathy: an unusual X-linked inheritance disorder. Hum Mutat. 2012;33(4):627-34.
- Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization and neurologic diversity. Genes Dev. 2000; 14:1169-80.
- Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protochaderin 19 mutation. Epilepsia. 2011;52(7):1251-7.
- Higurashi N, Shi X, Yasumoto S, et al. PCDH19 mutation in Japanese females with epilepsy. Epilepsy Res. 2012;99(1-2):28-37.
- Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet Syndrome but mainly affects females. PLoS Genet. 2009;5(2):e1000381.
- Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet Syndrome. Epilepsia. 2011;2:24-29.
- Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia. 2012; 53(12):2011-9.
- Higurashi N, Nakamura M, Sugai M, et al. PCDH19-related female-limited epilepsy: further details regarding early clinical features and therapeutic efficacy. Epilepsy Res. 2013;106(1-2):191-9.
- Lotte J, Bast T, Borusiak P, et al. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure. 2016; 35:106-10.
- Linee guida - Il trattamento dell’epilessia in età pediatrica. SINPIA, 2017.