




la Newsletter
Esordio acuto con coma iperammoniemico nei difetti del ciclo...
I difetti del ciclo dell’urea ad esordio tradivo acuto sono una...
Casi clinici n.1 e 2
Il signor A di anni 48, decede in iperammoniemia 72 ore dopo un intervento chirurgico alla colonna dorsale. Aveva goduto di buona salute fino all’anno prima quando, in concomitanza con l’insorgenza di dolori al dorso, aveva iniziato ad alimentarsi poco perdendo circa 10 kg. In una occasione si era presentato in Pronto Soccorso lamentando un peggioramento dei dolori e chiedendo un antidolorifico ed era apparso poco lucido e non ben orientato nel tempo. Era stato trattenuto in ospedale due giorni e trattato per via endovenosa con antidolorifico diluito in soluzione glucosata. Il lieve stato confusionale era stato considerato secondario al dolore ed era completamente scomparso alla dimissione.
.jpg) Le indagini sulla causa dell’iperammoniemia portano ad una diagnosi, che purtroppo giunge post-mortem, di difetto del ciclo dell’urea (Fig. 1) ed in particolare di deficit di ornitina transcarbamilasi (OTC). Gli esami biochimici per malattia metabolica mostrano citrullina plasmatica ridotta ed aumentata escrezione urinaria di acido orotico; l’analisi molecolare del gene OTC (che è localizzato sul cromosoma X) conferma successivamente la diagnosi con l’identificazione della variante patogena p.Ala208Thr (c.622G>A).
Le indagini sulla causa dell’iperammoniemia portano ad una diagnosi, che purtroppo giunge post-mortem, di difetto del ciclo dell’urea (Fig. 1) ed in particolare di deficit di ornitina transcarbamilasi (OTC). Gli esami biochimici per malattia metabolica mostrano citrullina plasmatica ridotta ed aumentata escrezione urinaria di acido orotico; l’analisi molecolare del gene OTC (che è localizzato sul cromosoma X) conferma successivamente la diagnosi con l’identificazione della variante patogena p.Ala208Thr (c.622G>A).
Circa un mese dopo, il signor B, fratello di A, di anni 36, in buona salute, giunge alla nostra osservazione chiedendo una consulenza genetica dopo aver ricevuto i risultati degli esami del fratello deceduto. Conosciuta l’anamnesi familiare, si spiega al soggetto di che tipo di malattia si tratti (genetica, metabolica a trasmissione X-linked, dove la femmina è in genere affetta con un fenotipo clinico più lieve del maschio) e quale sia il suo rischio di esserne affetto (50% se la madre è portatrice). Si esegue inoltre il prelievo per l’analisi di mutazione nota del gene OTC per verificare la presenza o assenza della mutazione familiare e, in attesa dei risultati, gli si consegna a scopo cautelativo, una “lettera di emergenza” che spiega la malattia e dà indicazioni per il trattamento di un eventuale episodio di scompenso acuto da deficit del ciclo dell’urea, da portare sempre con sé ed utilizzare in caso fosse anche lui affetto e presentasse uno scompenso metabolico acuto.
Circa 10 giorni dopo, il signor C, 43 anni, cugino per via materna di A e B, sempre stato in buona salute e noto per essere un avido mangiatore di carne, ha una grave lesione oculare e al volto in seguito ad un incidente domestico per il quale viene sottoposto a plurimi interventi in anestesia generale per avulsione dell’occhio destro e ricostruzione per piani delle ferite lacerocontuse presenti a livello di naso, mento, palpebra, labbro inferiore. È trattato con cortisone e antibiotici e si alimenta molto poco per le difficoltà ad assumere cibo per bocca. Tre giorni dopo il ricovero, il paziente diventa inizialmente aggressivo con i medici e con i familiari e successivamente entra in stato confusionale e coma lieve.
L’ammoniemia è 570 mcg/dl (vn fino a 80). Mentre i medici stanno cercando di capire le cause di questo quadro clinico, giunge in visita il cugino B, che comprende che probabilmente C soffre della stessa malattia di suo fratello A e consegna ai medici la propria lettera di emergenza che ha diligentemente tenuto nel portafoglio. Viene dunque iniziato il trattamento specifico (vedi sotto) per il deficit di OTC. L’ammoniemia ritorna alla norma in due giorni. Contemporaneamente sono eseguiti gli esami biochimici specifici che mostrano anche citrullina plasmatica bassa ed aumento dell’acido orotico urinario. Il paziente viene dimesso 15 giorni dopo l’episodio di iperammoniemia con dieta libera e trattamento con L-arginina per os. La diagnosi molecolare confermerà anche in lui la presenza della mutazione familiare nel gene OTC, mentre questa risulterà assente nel cugino B.
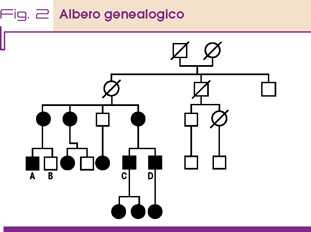 La famiglia è stata studiata per quanto possibile (Fig. 2) in relazione alla disponibilità dei singoli soggetti ed è stato identificato come affetto anche il signor D, fratello di C. Nella generazione precedente le due mamme di queste due coppie di cugini sono risultate affette, insieme ad una loro sorella che ha figli sani. Nella generazione successiva le bambine, figlie di C e D, sono risultate affette (eterozigoti obbligate in quanto hanno ereditato la X dal padre). Tutti questi individui identificati successivamente sono in buona salute e non hanno mai avuto disturbi alimentari o rifiuto di cibi proteici. Sono in follow-up con trattamento dietetico e farmacologico oppure solo in osservazione, in relazione ai risultati degli esami biochimici.
La famiglia è stata studiata per quanto possibile (Fig. 2) in relazione alla disponibilità dei singoli soggetti ed è stato identificato come affetto anche il signor D, fratello di C. Nella generazione precedente le due mamme di queste due coppie di cugini sono risultate affette, insieme ad una loro sorella che ha figli sani. Nella generazione successiva le bambine, figlie di C e D, sono risultate affette (eterozigoti obbligate in quanto hanno ereditato la X dal padre). Tutti questi individui identificati successivamente sono in buona salute e non hanno mai avuto disturbi alimentari o rifiuto di cibi proteici. Sono in follow-up con trattamento dietetico e farmacologico oppure solo in osservazione, in relazione ai risultati degli esami biochimici.
Caso clinico n. 3
37 anni, secondipara. Ottima salute in precedenza, nessun problema al primo parto che è stato fisiologico. In anamnesi un intervento chirurgico in anestesia generale senza problemi. Non riportata patologia epatica. Poche ore dopo il secondo parto sviluppa un disturbo comportamentale con alterazioni del controllo della vigilanza a cui si associano crisi comiziali. Esegue accertamenti neurologici (RMN encefalo e angiorisonanza) escludendo problematiche parenchimali di rilievo. Esame del liquor negativo per segni di infiammazione. Gli esami ematici rivelano iperammoniemia (416 mcg/dl) e ipertransaminasemia (ALT 823 U/l, AST 487 U/l). Si inizia emodiafiltrazione continua e nel contempo si cerca una diagnosi. Lo studio della situazione da parte dello staff medico porta alla ricerca di malattie metaboliche e si inviano al laboratorio di riferimento gli esami specifici: aminoacidi plasmatici e urinari, acido orotico urinario, acidi organici urinari. I risultati mostrano citrullina molto elevata, arginina ai limiti inferiori. Restanti esami nella norma.
Viene, quindi, formulata la diagnosi di difetto di argininosuccinato sintetasi (AS) o citrullinemia tipo 1 (Fig. 1), uno dei difetti del ciclo dell’urea, a trasmissione autosomica recessiva. Questa diagnosi è confermata successivamente dall’analisi molecolare del gene ASS1 che mostra due varianti patogene (p.Arg100Cys/p.Gly390Arg). La paziente è trattata con L-arginina cloridrato per ev e successivamente per os, e dieta ipoproteica. L‘emodiafiltrazione viene sospesa dopo 48 ore senza rebound di iperammoniemia. Alla prima visita di controllo la signora racconta che fin da bambina non aveva mai gradito la carne e da sempre ne assumeva pochissima, ma nell’ultima gravidanza, per riscontro di anemia ferrocarenziale, si era sforzata di assumerla, nonostante i suoi gusti alimentari.
Commento
Questi sono 3 casi di esordio acuto in età adulta di deficit del ciclo dell’urea. Sono solo una piccola parte dei casi analoghi visti in 30 anni nel nostro centro metabolico. Storie stupefacenti se non si conoscono queste malattie, e davvero difficili da riconoscere: non ci si spiega perché una persona che ha avuto una vita normale fino all’età adulta possa, all’improvviso, avere uno scompenso metabolico e andare in coma iperammoniemico a causa di una malattia genetica.
Siamo soliti pensare che le malattie genetiche si manifestano alla nascita, oppure dalla nascita in poi in modo progressivo. Invece in questo caso ci troviamo di fronte a malattie genetiche, ereditarie, che causano un malfunzionamento del ciclo dell’urea e che si possono anche manifestare in età adulta o adolescenziale, in modo acuto e drammatico, in occasione di stress catabolico acuto.
È questo il motivo per cui abbiamo voluto segnalare questi casi. A fronte di pazienti che riescono ad uscirne favorevolmente sappiamo che ce ne sono stati altri che hanno avuto una diagnosi post-mortem, come il signor A e anche, verosimilmente, altri pazienti morti senza diagnosi.
Cenni sui difetti del ciclo dell’urea
Il ciclo dell’urea è la sede di una serie di reazioni enzimatiche, che avvengono esclusivamente nel fegato, e permettono di trasformare l’ammoniaca, prodotto finale del catabolismo degli aminoacidi e tossica per l’organismo, in urea, non tossica, che viene escreta con le urine.
Per tutti gli enzimi del ciclo dell’urea (Fig. 1) sono stati riportati pazienti con un deficit: N-acetilglutammato sintetasi (NAGS), carbamilfosfato sintetasi (CPS), ornitina transcarbamilasi (OTC), argininosuccinato sintetasi (ASS), argininosuccinato liasi (ASL), arginasi (AS). La sindrome HHH (iperornitinemia, iperammonemia e omocitrullinuria) e la intolleranza alle proteine con lisinuria (IPL), sono dovute a difetti di trasporto di aminoacidi che partecipano al ciclo dell’urea (ornitina per la sindrome HHH e lisina, ornitina e arginina per IPL) e presentano sintomi simili pur non essendo definibili come difetti del ciclo dell’urea.
Si stima che i difetti del ciclo dell’urea abbiano complessivamente una frequenza di circa 1:20.000-30.000 nati. Approssimativamente si calcola che circa 2/3 dei pazienti abbiano un esordio grave, neonatale e circa 1/3 un esordio tardivo. Le forme che esordiscono tardivamente, a qualunque età, hanno un’attività enzimatica residua che permette un funzionamento appena sufficiente del ciclo in condizioni normali ma non è in grado di affrontare un “superlavoro” nelle situazioni di ipercatabolismo. Abbiamo visto che i pazienti a volte tendono a difendersi istintivamente dall’eccesso proteico, rifiutando la carne, o hanno saltuariamente qualche piccolo sintomo che passa inosservato. Abbiamo però anche visto che alcuni, come il signor C, in condizioni di benessere non hanno alcun sintomo e non tendono neppure a rifiutare i cibi proteici (il signor C era un avido mangiatore di carne).
Come e quando pensarci? Come impostare il trattamento immediato?
Si può osservare prima di tutto che sempre c’è una causa scatenante (forte stress catabolico dato da malattie intercorrenti, interventi chirurgici, diete dimagranti drastiche, parto, digiuno, terapie steroidee prolungate) che induce ipercatabolismo proteico e quindi forza l’attività del ciclo dell’urea che deve accelerare il passo per degradare l’ammoniaca e trasformarla in urea. A volte vi è anche associato un aumentato introito proteico alimentare, “forzato” da parte del paziente (es. pasti non usuali in occasione di feste, cambio dell’usuale regime alimentare per motivi diversi etc...).
Spesso il paziente, prima di andare in coma manifesta segni psichiatrici come un comportamento bizzarro o non usuale, improvvisi cambiamenti di umore, aggressività immotivata nei confronti dei familiari o di estranei. Di frequente i familiari successivamente raccontano che il paziente “sembrava un'altra persona”.
Nell’anamnesi personale del paziente si possono trovare piccoli indizi: rifiuto di assumere proteine animali fin dall’infanzia, frequenti cefalee, episodi di modificazione del comportamento nel corso della vita, che poi scomparivano senza apparente motivo dopo alcuni giorni.
Con i primi esami ematochimici in mano (ammoniemia alta, sintesi epatica nella norma, associata a possibile modesta citolisi) qualunque medico si dovrebbe rendere conto che non si tratta di iperammoniemia da epatopatia generalizzata o sue conseguenze (cirrosi, shunt porto-cavale, sanguinamento da varici esofagee…). Spesso però la situazione di urgenza, la necessità di decidere subito un trattamento, possono portare anche i migliori di noi a conclusioni avventate.
Sapere che esistono difetti genetici del ciclo dell’urea che si presentano in questo modo può aiutare a orientarsi più in fretta. Il ragionamento è: se il mio paziente non ha una patologia epatica, primaria o secondaria, che giustifica la sua iperammoniemia, è ragionevole pensare ad un difetto congenito del ciclo dell’urea che porta ad iperammoniemia pressoché isolata (solo transaminasi talvolta mosse con pseudocolinesterasi, albumina, prealbumina, protidemia nella norma).
Con questo sospetto si possono avviare gli esami specifici come il dosaggio degli aminoacidi plasmatici ed urinari e dell’acido orotico urinario (che richiedono alcuni giorni per la risposta) ma soprattutto iniziare, SUBITO, prima dell’arrivo dei risultati di questi esami, il trattamento d’urgenza come segue:
- somministrare sufficienti calorie per evitare il catabolismo endogeno (necessario un accesso centrale per nutrizione parenterale)
- l’apporto calorico, per os o per via ev secondo le condizioni del paziente, deve essere aproteico, cioè senza proteine e aminoacidi
- controindicata la somministrazione degli aminoacidi ramificati
- valutare se necessaria emodiafiltrazione in base alle condizioni del paziente e/o alla sua risposta alle prime ore di terapia disintossicante
- contemporaneamente contattare il centro per le malattie metaboliche più vicino e chiedere indicazioni riguardo al trattamento e alla possibile somministrazione di farmaci “scavenger”, cioè farmaci che permettono di eliminare molecole di azoto attraverso vie alternative al ciclo dell’urea, riducendo quindi il “carico” del ciclo dell’urea.
Esami diagnostici
Gli esami biochimici necessari per la diagnosi sono l’aminoacidogramma plasmatico, l’acido orotico e gli acidi organici urinari. In generale, in presenza di un difetto del ciclo dell’urea, il metabolita a monte del deficit è elevato nel plasma e quello a valle è ridotto. Come mostra la figura 1, il deficit di OTC ostacola la produzione di citrullina che è spesso bassa (ma può essere normale) in questa malattia; il carbamilfosfato, non potendo essere metabolizzato da OTC segue una via secondaria che porta alla produzione di acido orotico. L’acido orotico urinario è sempre elevato durante uno scompenso nel paziente con deficit di OTC.
Nella situazione del deficit di ASS o citrullinemia, avremo invece il marker della citrullina che risulterà elevata.
In un secondo momento, l’analisi genetica dimostrerà la presenza di varianti patogenetiche nei rispettivi geni relativi ai difetti del ciclo dell’urea negli individui affetti.
Sarà poi necessario indagare anche le famiglie dei pazienti. La citrullinemia è trasmessa con modalità autosomica recessiva ed entrambi i genitori dovrebbero risultare portatori sani di una delle due varianti genetiche identificate nel paziente; vanno indagati anche fratelli o sorelle del probando che potrebbero pure essere affetti e per il momento asintomatici. Nel caso invece del deficit di OTC, con trasmissione X-linked, l’indagine familiare è più complicata perché, oltre al genitore portatore (ma in questo caso comunque da considerare affetto) e ai fratelli, l’indagine va allargata, studiando l’albero genealogico, a tutti i parenti che potrebbero aver ricevuto il cromosoma X con il gene affetto. Ciò è importante perché la loro identificazione presintomatica potrebbe evitare uno scompenso acuto con rischio di vita. Esistono però anche casi con mutazione “de novo”, cioè presente nell’individuo affetto ma non nei genitori.
Da segnalare, infine, che lo screening neonatale esteso permette ora di identificare i casi di citrullinemia ad esordio tardivo. Sono perciò stati riconosciuti anche in Italia negli ultimi anni neonati con citrullinemia asintomatici, ma con le alterazioni biochimiche della malattia.
La presa in carico in un ambulatorio metabolico dalla nascita scongiura o almeno riduce il rischio di scompenso acuto nelle età successive. Si corre però in questi casi il rischio di sovra-trattamento, con diete drastiche o farmaci non necessari che possono indurre patologie iatrogene (prima di tutto inappetenza).
Conclusioni
I difetti del ciclo dell’urea ad esordio tradivo acuto sono una realtà clinica molto spesso misconosciuta. Nel caso di un paziente con iperammoniemia inspiegata, senza precedente malattia epatica, sono la diagnosi più probabile, anche se malattie rare. Il trattamento acuto è principalmente basato sulla somministrazione di calorie non proteiche per arrestare il catabolismo endogeno ed eventuale precoce emodialisi. La tempestività del trattamento è fondamentale per un buon esito clinico. Il contatto più precoce possibile con il centro per le malattie metaboliche è fondamentale per definire la terapia e il suo proseguimento dopo le prime 24-48 ore. Lo screening neonatale esteso identifica alcuni difetti del ciclo dell’urea (tra cui la citrullinemia) ma non tutti (il deficit di OTC è escluso).
Bibliografia
- Batshaw ML, Tuchman M, Summar M, et al. A longitudinal study of urea cycle disorders. Mol Genet Metab. 113(1-2):127-30, 2014.
- Deignan JL, Cederbaum SD, Grody WW. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab. 93(1):7-14, 2008.
- Haberle J, Boddaert N, Burlina A et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphaned Journal Rare Diseases 7: 32, 2012.
- Helman G, Pacheco-Colón I, Gropman AL. The urea cycle disorders. Semin Neurol. 34(3):341-9, 2014.
- IPERAMMONIEMIE EREDITARIE; INTOLLERANZA ALLE PROTEINE CON LISINURIA E SINDROME HHH. Su: https://www.regione.lombardia.it ricerca libera digitare Rete Regionale per le malattie rare- Regione Lombardia.> Link a centro di Coordinamento della rete regionale per le malattie rare > percorsi diagnostici, terapeutici e assistenziali (PDTA). Ultimo accesso il giorno 8 settembre 2019.
- Matsumoto S, Häberle J, Kido J, et al. Urea cycle disorders-update. J Hum Genet. 64(9):833-847, 2019.