




la Newsletter

Laura Romaggioli1, Giuseppina Cusano1, Isabella Quinti2, Cinzia Milito2
1Centro di Riferimento Regionale per le Immunodeficienze Primitive, Azienda Ospedaliera-Universitaria Policlinico Umberto I, Roma; 2Dipartimento di Medicina Molecolare, Sapienza Università di Roma
Immunodeficienza comune variabile | L’immunodeficienza comune...
L’immunodeficienza comune variabile comprende un gruppo...
L’immunodeficienza comune variabile (ICV) è la più frequente immunodeficienza primitiva sintomatica, con una prevalenza di 1:25.000. Comprende un gruppo eterogeneo di disordini immunitari caratterizzati da ipogammaglobulinemia, aumentata suscettibilità alle infezioni e un ampio spettro di comorbidità. Colpisce adulti e bambini con un picco di insorgenza dei sintomi tra la prima e la terza decade e una distribuzione simile nei due sessi. L’eterogeneità e il diverso grado di severità di manifestazioni cliniche rendono difficile una diagnosi precoce, che è solitamente posta con un ritardo medio di 5 anni.
Eziopatogenesi
Il maggior numero di casi è sporadico; il 10-25% dei casi è familiare, con un’ereditarietà autosomica dominante. In alcuni pazienti è stata identificata una mutazione di un gene coinvolto nella maturazione e differenziazione dei linfociti B (TACI, BAFF-R, CD19). I meccanismi patogenetici non sono ad oggi ancora completamente noti. Alcuni pazienti hanno un numero normale di linfociti B circolanti con alterazioni della differenziazione terminale o della differenziazione in linfociti di memoria e/o plasmacellule. Indipendentemente dal meccanismo patogenetico sottostante, il difetto comune è l’insufficiente produzione anticorpale.
Quadro clinico
L’ICV presenta un’ampia varietà fenotipica: da forme scarsamente sintomatiche a quadri molto severi, con estrema suscettibilità alle infezioni, manifestazioni autoimmuni, immunodisregolazione, disordini linfoproliferativi e neoplasie.
Complicanze infettive
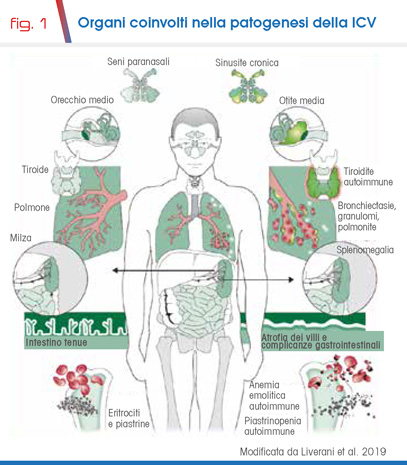 Il quadro clinico può essere caratterizzato da infezioni ricorrenti e gravi del tratto respiratorio superiore ed inferiore (sinusiti, otiti, bronchiti e polmoniti), da batteri capsulati (S. pneumoniae, H. influenzae). Come complicanza delle infezioni, è frequente il riscontro di bronchiectasie e di deficit respiratori ostruttivi e restrittivi. Particolarmente frequenti sono le infezioni del tratto gastrointestinale, da virus e parassiti, quali Giardia lamblia e Campylobacter jejuni. Sono anche descritte infezioni da patogeni atipici, quali Mycoplasma spp e Ureaplasma urealyticum. Le infezioni da patogeni opportunisti (Pneumocystis jirovecii) non sono tipiche e devono far sospettare un associato deficit dell’immunità cellulare o una diagnosi alternativa (Fig.1).
Il quadro clinico può essere caratterizzato da infezioni ricorrenti e gravi del tratto respiratorio superiore ed inferiore (sinusiti, otiti, bronchiti e polmoniti), da batteri capsulati (S. pneumoniae, H. influenzae). Come complicanza delle infezioni, è frequente il riscontro di bronchiectasie e di deficit respiratori ostruttivi e restrittivi. Particolarmente frequenti sono le infezioni del tratto gastrointestinale, da virus e parassiti, quali Giardia lamblia e Campylobacter jejuni. Sono anche descritte infezioni da patogeni atipici, quali Mycoplasma spp e Ureaplasma urealyticum. Le infezioni da patogeni opportunisti (Pneumocystis jirovecii) non sono tipiche e devono far sospettare un associato deficit dell’immunità cellulare o una diagnosi alternativa (Fig.1).
Complicanze non infettive
Le complicanze infiammatorie non infettive quali manifestazioni autoimmuni, malattia polmonare, linfoproliferazione e neoplasie sono presenti nel 70% dei casi. Spesso si associano ad una prognosi più grave e a una ridotta qualità di vita.
Autoimmunità
In un terzo dei pazienti, le manifestazioni autoimmuni costituiscono il sintomo d’esordio. Le più comuni sono le citopenie quali porpora trombocitopenica idiopatica, anemia emolitica autoimmune e neutropenia autoimmune. È inoltre riportata l’associazione con altre patologie autoimmuni quali artriti, uveiti, vasculiti, tiroiditi, vitiligo e cirrosi biliare primitiva.
Coinvolgimento gastrointestinale ed epatico
La frequenza delle manifestazioni gastrointestinali è variabile (20-60%). I sintomi gastrointestinali includono gastrite atrofica, anemia perniciosa, iperplasia nodulare linfoide, diarrea cronica e celiachia. Alcuni pazienti sviluppano un’enteropatia cronica, di tipo infiammatorio, con malassorbimento. Non è infrequente l’ipertensione portale non cirrotica da iperplasia nodulare rigenerativa del fegato.
Malattia granulomatosa sistemica
La malattia granulomatosa colpisce il 10-20% dei pazienti, coinvolge diversi organi (fegato, milza) e si associa a manifestazioni autoimmuni. Istologicamente i granulomi non caseosi somigliano a quelli della sarcoidosi. A livello polmonare si associano ad infiltrati linfoidi determinando la “malattia granulomatosa linfocitica polmonare interstiziale” (granulomatous lymphocytic interstitial lung disease, GLILD). Ad oggi, la patogenesi della GLILD non è completamente chiarita ma sembra riconducibile ad una condizione di immunodisregolazione. La GLILD determina un rapido decadimento della funzionalità respiratoria ed è associata ad una prognosi clinica peggiore.
Neoplasie
Rispetto alla popolazione generale, nei pazienti con ICV è stata osservata un’aumentata prevalenza di neoplasie ematologiche e cancro gastrico.
Le neoplasie più comunemente diagnosticate sono i linfomi B e l’adenocarcinoma gastrico. In uno studio italiano su pazienti con ICV, il cancro gastrico è il secondo tumore diagnosticato in ordine di frequenza e la prima causa di morte.
Diagnosi
 I criteri diagnostici elaborati dall’ESID (European Society for Immunodeficiencies) nel 2019 permettono di porre diagnosi di ICV in soggetti di età > 4 anni se presentano almeno una delle seguenti condizioni cliniche:
I criteri diagnostici elaborati dall’ESID (European Society for Immunodeficiencies) nel 2019 permettono di porre diagnosi di ICV in soggetti di età > 4 anni se presentano almeno una delle seguenti condizioni cliniche:
- aumentata suscettibilità ad infezioni, manifestazioni autoimmuni, malattia granulomatosa, linfoproliferazione policlonale, familiarità per difetti anticorpali;
- marcata riduzione di IgG e IgA e/o bassi livelli sierici di IgM (in due misurazioni, con valori < a due deviazioni standard rispetto al valore normale per fascia di età);
- scarsa risposta anticorpale ai vaccini (e/o assenza di isoemoagglutinine).
Per porre diagnosi vanno escluse altre cause di ipogammaglobulinemia e difetti dei linfociti T.
Elevati livelli di IgE o IgM devono orientare verso altri tipi di immunodeficienze o fenomeni di disregolazione del sistema immunitario (Tab.1).
Terapia
Terapia sostitutiva con immunoglobuline
La terapia sostitutiva con immunoglobuline riduce la mortalità, l’incidenza delle complicanze infettive e contribuisce al miglioramento della qualità di vita. Attualmente, le immunoglobuline possono essere somministrate per via endovenosa (IVIG), sottocutanea (SCIG) e sottocutanea facilitata (fSCIG). Le IVIG sono di solito ben tollerate, tuttavia, effetti collaterali sistemici (febbre, cefalea, vomito, mialgia, dolore addominale) possono manifestarsi sia in acuto che dopo ore dall’infusione. Le reazioni moderate acute e ritardate possono essere prevenute con l’utilizzo di corticosteroidi pre- e/o post-infusione. Le reazioni anafilattiche severe sono molto rare. La somministrazione sottocutanea è in genere ben tollerata e può essere effettuata a domicilio; può associarsi ad eventi avversi di breve durata in sede di inoculo (eritema ed edema). Il dosaggio di IgG sostitutive varia tra 300-600 mg/kg ogni 3-4 settimane con l’obiettivo di mantenere i livelli plasmatici di IgG a valori >500 mg/dl, per ridurre il rischio infettivo. È necessario individualizzare la terapia in pazienti con comorbidità quali bronchiectasie, enteropatia e splenomegalia che richiedono dosaggi più alti per rimanere liberi da infezioni. La scelta della via di somministrazione va concordata con il paziente e sulla base della disponibilità di accessi venosi, patologie concomitanti e compliance del paziente. Le attuali vie di somministrazione presentano un’efficacia sovrapponibile nel prevenire gli episodi infettivi. Recenti studi clinici dimostrano che le SCIG hanno migliorato la qualità di vita dei pazienti grazie alla gestione domiciliare della terapia.
Terapia delle infezioni
Sebbene la terapia sostitutiva riduca il rischio infettivo, è spesso necessario ricorrere a terapia antimicrobica per prevenire le complicanze. Le infezioni ricorrenti delle alte vie respiratorie esitano in sinusiti croniche per le quali è utile la profilassi antibiotica con macrolidi o amoxicillina. Nei pazienti con insufficienza respiratoria e bronchiectasie, l’uso di corticosteroidi e broncodilatatori e la fisiokinesiterapia respiratoria possono prevenire o rallentare la progressione del danno respiratorio. Un recente trial randomizzato di fase II ha evidenziato che l’utilizzo di profilassi antibiotica con macrolidi a basse dosi sia efficace nel prevenire le riacutizzazioni polmonari.
Terapia delle manifestazioni autoimmuni e granulomatose
Le terapie utilizzate ad oggi non incidono sulle complicanze autoimmuni e infiammatorie, che contribuiscono significativamente alla morbidità e mortalità dei pazienti. Nei pazienti con citopenia autoimmune è indicato l’utilizzo di steroidi a dosaggi standard (1-2 mg/kg/die) mentre nei casi refrattari vi è indicazione all’uso di rituximab. Nei pazienti con coinvolgimento intestinale vengono prescritti mesalazina e budesonide in associazione a basse dosi di steroidi. Nei pazienti con quadri infiammatori severi o refrattari può essere utilizzato infliximab. Nei pazienti con malattia granulomatosa il trattamento è empirico; possono essere usati corticosteroidi ad alte dosi mentre immunosoppressori quali azatioprina, micofenolato o ciclosporina hanno dimostrato effetti variabili. L’utilizzo di rituximab nella GLILD migliora il danno e la funzione polmonare.
Prognosi
Le comorbidità non infettive peggiorano la prognosi dei pazienti. La sopravvivenza globale è migliorata significativamente dopo l’introduzione della terapia sostitutiva. Ad oggi, la sopravvivenza stimata è di oltre il 50% a 40 anni dalla diagnosi, in particolar modo nei pazienti che non sviluppano neoplasie.
Bibliografia
- Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract. 2019;7(6):1763-1770.
- Ameratunga R, Allan C, Woon ST. Defining Common Variable Immunodeficiency Disorders in 2020. Immunol Allergy Clin North Am. 2020;40(3):403-420.
- Bonilla FA. Personalized therapy for common variable immunodeficiency. Allergy Asthma Proc. 2020;41(1):19-25.
- Gupta S, Pattanaik D, Krishnaswamy G. Common Variable Immune Deficiency and Associated Complications. Chest. 2019;156(3):579-593.
- Milito C, Pulvirenti F, Cinetto F, et al. Double-blind, placebo-controlled, randomized trial on low-dose azithromycin prophylaxis in patients with primary antibody deficiencies. J Allergy Clin Immunol. 2019;144(2):584-593.e7.
- Ng J, Wright K, Alvarez M, et al. Rituximab Monotherapy for Common Variable Immune Deficiency-Associated Granulomatous-Lymphocytic Interstitial Lung Disease. Chest. 2019;155(5):e117-e121.