




la Newsletter
Infezioni respiratorie ricorrenti: quando sospettare la discinesia...
Le infezioni respiratorie ricorrenti, causate per lo più da...
Le infezioni respiratorie ricorrenti, causate per lo più da virus, ma anche da batteri, possono interessare fino al 25% dei bambini sotto l'anno di età e il 18% dei bambini di età compresa tra 1 e 4 anni (1) e rappresentano pertanto una sfida diagnostica pressoché quotidiana per il pediatra che deve dirimere se una rinite persistente o un'otite media secretiva oppure una bronchite catarrale, problemi comuni nei bambini in età pre-scolare, siano sottesi o meno da una patologia genetica o da uno stato di immunodeficienza, primaria o secondaria.
Quando si parla di infezioni respiratorie ricorrenti si fa riferimento ad una diagnosi di 8 o più episodi infettivi respiratori per anno nel bambino di età minore di 3 anni oppure di 6 o più episodi infettivi respiratori nel bambino con più di 3 anni di età; questi criteri possono essere lievemente differenti se varia il sito di infezione interessato (2). Nell'adulto tale problema è meno frequentemente riscontrabile, a meno che non vi siano delle comorbilità che favoriscono la recidiva degli episodi infettivi.
Diagnosi differenziale
.jpg) Quando il pediatra o lo specialista pneumologo oppure l'internista si trovano a dover far luce eziologicamente su questo problema, devono anzitutto effettuare una diagnosi differenziale tra diverse patologie che possono manifestarsi con aspetti clinici comuni e devono richiedere l'esecuzione di esami specifici (Tab. 1).
Quando il pediatra o lo specialista pneumologo oppure l'internista si trovano a dover far luce eziologicamente su questo problema, devono anzitutto effettuare una diagnosi differenziale tra diverse patologie che possono manifestarsi con aspetti clinici comuni e devono richiedere l'esecuzione di esami specifici (Tab. 1).
La presenza di una ricorrenza infettiva a carico delle vie respiratorie impone una diagnosi differenziale in primo luogo con le sindromi da immunodeficienza e la Fibrosi Cistica (FC), oppure può essere sostenuta da anomalie anatomiche a carico dell'albero respiratorio; inoltre vanno indagati un eventuale deficit di alfa-1-antitripsina o la possibile presenza di asma bronchiale e di reflusso gastro-esofageo.
Una volta effettuate le indagini di primo livello, e se queste risultano negative, si può pensare ad una patologia più rara quale la Discinesia Ciliare Primaria (PCD), che presenta una diagnostica più complessa e sofisticata. La diagnosi viene derivata in base a sintomi, segni e risultati di esami dedicati, considerati nel loro insieme, poiché non esiste una singola caratteristica specifica della malattia.
Epidemiologia
La PCD è una malattia "rara" con una prevalenza di 1: 20.000, in cui l'attività ciliare è anomala: essa è caratterizzata dalla presenza di infezioni ricorrenti dell'albero respiratorio, sia a carico delle alte che delle basse vie, e nel 50% dei casi circa, è accompagnata da anomalie di posizione degli organi interni. Quando queste anomalie, definite "eterotassie", sono presenti, è ovviamente più facile porre il sospetto diagnostico; nel caso invece in cui questa caratteristica non sia presente, si assiste spesso ad un ritardo diagnostico. L'età media della diagnosi si attesta sui 4 anni di età, anche se non mancano diagnosi effettuate in età adulta (3). La malattia è stata descritta per la prima volta nel 1933 da Kartagener (4) che ha riportato l'osservazione di 4 pazienti con la triade: sinusite, bronchiettasie e situs inversus (posizione degli organi speculare rispetto a quella fisiologica). Si deve tuttavia ad Afzelius, nel 1976, il riconoscimento della presenza di un difetto ultrastrutturale a carico delle cilia (5). Poiché non sempre il situs inversus è presente, oggi si preferisce comprendere la sindrome nella denominazione Discinesia Ciliare Primaria.
Le ciliopatie
La malattia fa parte comunque di un gruppo eterogeneo di malattie ancora più ampio, chiamate genericamente ciliopatie ovvero dovute ad alterazioni delle cilia, soprattutto di quelle respiratorie, ma più raramente, anche di quelle a carico di altri organi ed apparati (6).
.jpg) La mucosa respiratoria che riveste l'intero apparato respiratorio, a partire dal naso fino alle più piccole diramazioni bronchiali è di tipo ciliato: le cellule ciliate rivestono tutta la superficie delle vie di conduzione dell'aria (Fig. 1), alternandosi alle cellule mucipare caliciformi (in rapporto di circa 5:1) che sono deputate alla secrezione del muco, unitamente alla quota più rilevante di muco secreta dalle ghiandole situate nella tonaca sottomucosa.
La mucosa respiratoria che riveste l'intero apparato respiratorio, a partire dal naso fino alle più piccole diramazioni bronchiali è di tipo ciliato: le cellule ciliate rivestono tutta la superficie delle vie di conduzione dell'aria (Fig. 1), alternandosi alle cellule mucipare caliciformi (in rapporto di circa 5:1) che sono deputate alla secrezione del muco, unitamente alla quota più rilevante di muco secreta dalle ghiandole situate nella tonaca sottomucosa.
Questi due componenti, cilia e muco, sono di fondamentale importanza per un corretto funzionamento del meccanismo di depurazione dell'aria respirata che è la clearance mucociliare: infatti le cilia sono in continuo movimento (circa 600-1200 battiti al minuto) per poter trasportare verso la faringe dove viene deglutito, il sottile film di muco che appoggia sulla loro sommità e che intrappola le sostanze nocive e le particelle inalate (velocità di trasporto mucociliare: 5-10 mm/ min.). In questo modo le vie aeree, al di sotto della trachea, vengono fisiologicamente mantenute sterili.
Genetica
La PCD è una malattia congenita, che viene prevalentemente ereditata in modo autosomico recessivo, benché esistano anche segnalazioni per un'ereditarietà di tipo dominante o legata al cromosoma X. Nel caso più frequente di ereditarietà autosomica recessiva, l'individuo che è affetto ha i due genitori che sono portatori dell'allele mutato (eterozigosi); l'individuo affetto ha una mutazione bi-allelica (omozigosi) e a sua volta ha una probabilità molto bassa di avere un figlio con la stessa malattia se il coniuge non è portatore della stessa mutazione. La malattia è più probabile nella progenie di relazioni tra consanguinei ed ha una probabilità 1:4 in ogni concepimento in cui i genitori sono entrambi portatori sani. Tuttavia, in questa sindrome vi è spesso anche un problema di infertilità nel maschio e di sub-fertilità nella donna, correlati ad un'alterata motilità degli spermatozoi e delle cilia delle tube uterine, rispettivamente.
Clinica
.jpg) I sintomi e i segni clinici della PCD variano in funzione dell'età e tendono ad aumentare con essa: la tabella 2 riporta i sintomi e segni più comuni e la loro relativa frequenza, mentre la tabella 3 riporta le manifestazioni più comuni suddivise secondo l'età.
I sintomi e i segni clinici della PCD variano in funzione dell'età e tendono ad aumentare con essa: la tabella 2 riporta i sintomi e segni più comuni e la loro relativa frequenza, mentre la tabella 3 riporta le manifestazioni più comuni suddivise secondo l'età.
Alla nascita possono essere presenti solo pochi sintomi come ad es. il distress respiratorio neonatale o alterazioni della lateralità degli organi, molte volte i sintomi cominciano a manifestarsi solo durante l'infanzia, mentre alcuni sintomi possono essere evidenti solo in età adulta, ad es. l'infertilità. Durante la gravidanza il riscontro di situs viscerum inversus impone futuri accertamenti al fine di confermare o escludere la diagnosi, poiché si stima che circa un quarto dei soggetti con tale anomalia sia affetto da PCD (7).
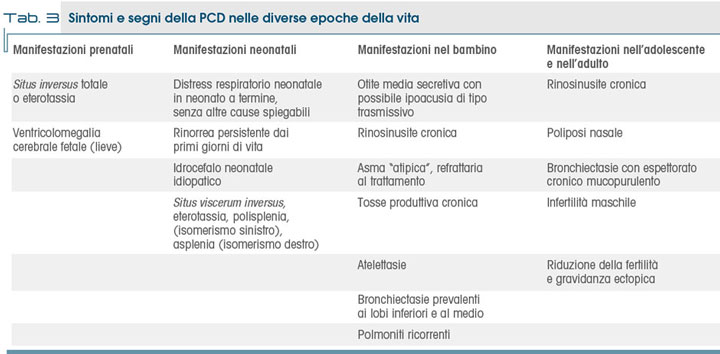
Durante il periodo neonatale tra le manifestazioni più frequenti vi sono il distress respiratorio alla nascita, non altrimenti spiegabile, con tachipnea e desaturazione arteriosa, che talora necessita di ventilazione meccanica, oppure la polmonite.
Più raramente, la presenza di malattie cardiache congenite, che più spesso accompagnano un situs viscerum incompleto o forme diverse di eterotassia, specialmente la trasposizione di grossi vasi, possono rappresentare una manifestazione precoce.
Una rinorrea cronica persistente è un altro frequente sintomo d'esordio, spesso già presente in epoca neonatale. La presenza di reflusso gastro-esofageo severo, talora con atresia esofagea è anche possibile, ma più rara.
Durante l'infanzia si evidenziano episodi infettivi ricorrenti a carico delle vie aeree superiori o inferiori: sinusiti, otiti, bronchiti, polmoniti oppure può essere presente una tosse cronica; talvolta si hanno manifestazioni asmatiche, refrattarie alle comuni terapie. Nell'adulto il quadro è sovente dominato da sinusite cronica, talora con poliposi nasale associata, da infezioni dell'albero bronchiale e da polmoniti ripetute, che portano allo sviluppo di bronchiectasie.
Le bronchiectasie sono prevalentemente di tipo cilindrico e a distribuzione segmentale, più spesso a carico del lobo medio, della lingula e dei segmenti basali: a volte queste sono già presenti in età pediatrica, più spesso si riscontrano in età adulta e si accompagnano a declino funzionale; un ispessimento delle pareti bronchiali è uno dei rilievi radiologici più precocemente riscontrabile.
Le manifestazioni cliniche della malattia sono simili a quelle della FC: la prognosi è comunque buona e il decorso nettamente più positivo rispetto alla FC. La funzione respiratoria, benché tenda a peggiorare con l'età, mostra decrementi minori rispetto alla FC, con una riduzione media del FEV1 pari a 0.8% rispetto al 3.6% riportato per la FC (8).
Indagine ultrastrutturale delle cilia
Mentre la FC è una malattia congenita correlata a modificazioni del film di muco per difettoso funzionamento del canale trans-membrana per il cloro (CFTR), la PCD è invece correlata ad alterazioni dell'ultrastruttura delle cilia respiratorie, prevalentemente all'assenza dei bracci esterni e/o di quelli interni di dineina che rappresenta la proteina contrattile che, in accoppiamento con l'ATP, è responsabile del movimento ciliare.
.jpg) Tale difetto è visibile al microscopio elettronico a trasmissione esaminando la sezione trasversale delle cilia, specialmente nella parte intermedia del cilio: questo esame è pertanto fondamentale per la diagnosi di PCD. La figura 2 mostra schematicamente la struttura del cilio in sezione trasversale, contenente al suo interno nove coppie periferiche di microtubuli ed una coppia centrale; le coppie periferiche presentano bracci esterni ed interni di dineina e sono connesse tra loro da legami di nexina; le coppie periferiche sono inoltre connesse alla coppia centrale attraverso i ponti radiali.
Tale difetto è visibile al microscopio elettronico a trasmissione esaminando la sezione trasversale delle cilia, specialmente nella parte intermedia del cilio: questo esame è pertanto fondamentale per la diagnosi di PCD. La figura 2 mostra schematicamente la struttura del cilio in sezione trasversale, contenente al suo interno nove coppie periferiche di microtubuli ed una coppia centrale; le coppie periferiche presentano bracci esterni ed interni di dineina e sono connesse tra loro da legami di nexina; le coppie periferiche sono inoltre connesse alla coppia centrale attraverso i ponti radiali.
.jpg) Nella figura 3 è possibile osservare l'aspetto ultrastrutturale del cilio normale (a sinistra) e nella PCD (a destra).
Nella figura 3 è possibile osservare l'aspetto ultrastrutturale del cilio normale (a sinistra) e nella PCD (a destra).
Studio della motilità ciliare
La diagnosi di PCD può essere tuttavia più complessa, perché oltre alle forme classiche in cui è presente tale deficit ultrastrutturale, possono esservi forme con alterazioni ultrastrutturali meno comuni (Tab. 4) o addirittura, rari casi senza alterazioni ultrastrutturali, ma caratterizzati solamente da alterazioni funzionali, ovvero unicamente da una riduzione dell'efficienza del battito ciliare.

.jpg) Complemento dell'indagine ultrastrutturale delle cilia respiratorie è pertanto lo studio della motilità ciliare, che viene effettuato solitamente utilizzando cellule ciliate di provenienza dalla mucosa nasale, prelevate mediante uno "spazzolamento" della mucosa del turbinato nasale inferiore e procedendo nel giro di pochi minuti all'osservazione ex vivo del movimento ciliare (Fig. 4).
Complemento dell'indagine ultrastrutturale delle cilia respiratorie è pertanto lo studio della motilità ciliare, che viene effettuato solitamente utilizzando cellule ciliate di provenienza dalla mucosa nasale, prelevate mediante uno "spazzolamento" della mucosa del turbinato nasale inferiore e procedendo nel giro di pochi minuti all'osservazione ex vivo del movimento ciliare (Fig. 4).
Naturalmente, tale esame può essere effettuato anche sulle cellule della mucosa bronchiale, qualora sia già programmata l'esecuzione di una fibrobroncoscopia: tale evenienza meno comune, può rendersi necessaria in caso di mancato reperimento di cellule ciliate a livello nasale, dopo aver ripetuto il brushing in diverse occasioni. Con questo esame si ricava un'informazione sulla frequenza del battito ciliare (valori normali medi: 10- 12 Hz) e sulla qualità del movimento (se coordinato e flessuoso come di norma, oppure rigido o assente). Negli anni più recenti si è affermata una tecnica di indagine che prevede lo studio della motilità con telecamera ad alta velocità in modo da poter visualizzare in modo più dettagliato il movimento delle cilia "frame by frame". Spesso i pazienti con PCD hanno un movimento ciliare molto ridotto o addirittura assente nella maggior parte dei campi esaminati al microscopio, anche se possono esservi casi in cui il movimento ciliare appare addirittura iperfrequente, ma il movimento non risulta corretto e non consente un efficiente trasporto del film di muco. Le cilia in cui mancano le braccia di dineina sono quelle che mostrano le alterazioni più marcate della motilità e rappresentano circa il 90% delle PCD (9); esse hanno due tipi di movimento asincrono multiplanare: rotazionale "a frullino" oppure "vibrazionale". Invece le cilia in cui mancano i ponti radiali hanno un aspetto afflosciato, con movimento rotatorio a "cavatappi" in due punti dell'assonema, mentre le cilia con difetto di traslocazione mostrano un movimento di flessione della sola parte apicale dell'assonema, mentre la base rimane rigida, il movimento avviene su diversi piani benché la frequenza possa essere ancora normale. Poiché numerose malattie respiratorie di tipo acquisito si accompagnano ad alterazioni della clearance mucociliare, è necessario differenziare la PCD dai difetti ciliari di tipo secondario.
Il percorso diagnostico
I due esami sopracitati che fanno parte del complesso ed articolato percorso diagnostico di questa malattia rara, vengono solitamente preceduti dalla misurazione dell'ossido nitrico nasale che nella PCD ha caratteristicamente un valore molto basso: questo test si è affermato nell'ultimo decennio come test di screening per la PCD. Valori inferiori a 200 ppb nell'adulto e <100 ppb nel bambino sono sospetti per la malattia (10).
Tutti le indagini diagnostiche vanno effettuate in condizioni di benessere o comunque di stabilità clinica per evitare fattori confondenti: poiché come già accennato, lo studio della motilità ciliare non consente di differenziare le discinesie primarie da quelle secondarie, la diagnosi di PCD non può essere posta solamente attraverso il risultato di questa indagine. La diagnosi può derivare solo dall'attenta valutazione dei risultati ottenuti con le diverse metodiche e, sovente, in seguito al coinvolgimento di diverse figure specialistiche oltre al pediatra e al pneumologo, quali l'otorinolaringoiatra, l'audiologo, il genetista, il ginecologo (nell'adulto), l'anatomo-patologo, il radiologo.
 Va considerato anche che negli ultimi anni si è esteso notevolmente lo spettro clinico di queste malattie conosciute e raggruppate sotto il termine di "ciliopatie", per cui accanto ai sintomi e segni classici possono essere presenti alcuni meno frequenti o rari che però non vanno sottovalutati o tralasciati (Tab. 5): oggi sappiamo ad esempio, che le cilia possono essere presenti in quasi tutti gli organi del corpo umano, sebbene con caratteristiche differenti secondo la sede, che ci sono cilia "mobili" e cilia "non mobili", con prevalente funzione sensoriale, e conosciamo patologie in cui vi sono alterazioni combinate delle cilia mobili e non mobili (11).
Va considerato anche che negli ultimi anni si è esteso notevolmente lo spettro clinico di queste malattie conosciute e raggruppate sotto il termine di "ciliopatie", per cui accanto ai sintomi e segni classici possono essere presenti alcuni meno frequenti o rari che però non vanno sottovalutati o tralasciati (Tab. 5): oggi sappiamo ad esempio, che le cilia possono essere presenti in quasi tutti gli organi del corpo umano, sebbene con caratteristiche differenti secondo la sede, che ci sono cilia "mobili" e cilia "non mobili", con prevalente funzione sensoriale, e conosciamo patologie in cui vi sono alterazioni combinate delle cilia mobili e non mobili (11).
Per questi motivi, il quadro classico finora descritto, può essere più complesso, con la presenza ad es. di idrocefalo o di retinite pigmentosa, o di rene policistico oppure di atresia biliare e cisti epatiche, a testimonianza del coinvolgimento delle cellule ciliate non mobili.
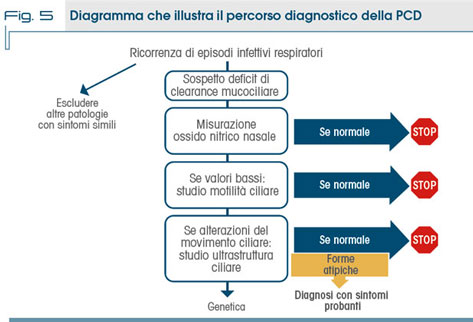 Nella figura 5 è riassunto il percorso diagnostico della PCD.
Nella figura 5 è riassunto il percorso diagnostico della PCD.
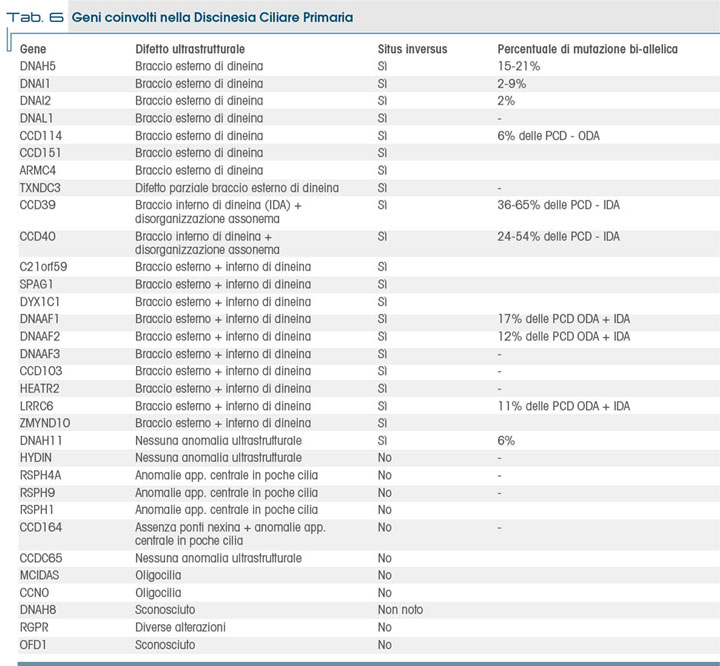
Anche dal punto di vista genetico, la malattia è eterogenea. L'esame genetico può essere effettuato, anche se la correlazione del gene mutato con la malattia è meno stretta rispetto ad esempio, alla FC. Ad oggi, si conoscono 32 geni che possono essere coinvolti nella PCD (12); considerato tuttavia che le cilia contengono almeno 200 differenti proteine, ciascuna codificata da geni separati, e che vi sono geni che codificano anche per l'assemblaggio e la regolazione delle cilia, si comprende come la malattia sia molto complessa dal punto di vista genetico ed il numero dei possibili geni candidati sia ampio. Alcuni di questi geni codificano per le catene pesanti della dineina e i loro siti di fosforilazione, altri per le tubuline che formano i microtubuli e altri per le proteine che legano le catene pesanti della dineina alle tubuline. In tabella 6 sono riportati i geni descritti in letteratura come possibili responsabili di ciliopatia.
Ringraziamenti
Si ringrazia il Dr. Luigi Ghilardini per l'assistenza tecnica nell'elaborazione delle immagini.
Bibliografia
- De Martino M, Ballotti S. The child with recurrent respiratory infections: normal or not? Pediatr Allergy Immunol. 2007; 18: 13-18.
- Bush A. Recurrent respiratory infections. Pediatr Clin North Am. 2009; 56 (1): 67-100.
- Coren ME, Meeks M, Morrison I, et al. Primary ciliary dyskinesia: age at diagnosis and symptom history. Acta Paediatrica 2002; 91: 667-669.
- Kartagener M. Zur pathogenese der bronkiectasien bei situs viscerum inversus. Beitr Klin Tuberk Spezif Tuberkuloforsch,1933; 83: 489-501.
- Afzelius BA. A human syndrome caused by immotile cilia. Science 1976; 193: 317-319.
- Brown JM, Witman GB. Cilia and disease. Bioscience 2014; 64: 1126-1137.
- Hogg C. Primary ciliary dyskinesia: when suspect the diagnosis and how to confirm it. Paediatr Resp Rev 2009; 10: 44-50.
- Noone PG, Leigh MW, Sannuti A, et al. Primary ciliary dyskinesia. Diagnostic and phenotypic features. AJRCCM 2004; 169:459-467.
- Chilvers MA, Rutman A, O'Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J All Clin Immunol 2003; 112: 518-523.
- Leigh MW, Hazucha MJ, Chawla KK, et al. Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann Am Thorac Soc 2013; 10 (6): 574-581.
- Horani A, Ferkol TW. Primary ciliary dyskinesia and associated sensory ciliopathies. Expert Rev Respir Med 2016; 10 (5): 569-576.
- Daniels ML, Noone PG. Genetics, diagnosis and future treatment strategies for primary ciliary dyskinesia. Expert Opin Orphan Drugs 2015,;3 (1): 31-44.
- Kurkowiak M, Zietkiewicz E, Witt M. Recent advances in ciliary dyskinesia genetics. J Med Genet 2015; 52: 1-9.
- Shapiro AJ, Zariwala MA, Ferkol T, et al. Diagnosis, monitoring, and treatment recommendations based on state of the art review. Pediatr Pulmonol 2016; 51 (2): 115-132.