




la Newsletter

Alberto Piperno1,2, Mara Botti1, Raffaella Mariani1
1Fondazione IRCCS San Gerardo, SSD Malattie Rare, Monza; European Reference Network (ERN) – Euro BloodNet; 2Centro Ricerca Tettamanti, Monza
La diagnosi genetico-molecolare dell’emocromatosi HFE-correlata...
Il test genetico molecolare ha rilevanza per confermare la...
L’emocromatosi (HC) HFE-correlata (HFE-HC) è la forma più frequente di HC nelle popolazioni di origine europea. Altri geni (HJV, HAMP, TFR2, SLC40A1) sono responsabili di forme rare o ultra-rare di HC (1,2). In questo contesto eterogeneo, il test genetico molecolare ha una rilevanza critica non solo per confermare la diagnosi, ma anche per prevenire le complicanze della patologia nei soggetti asintomatici.
Fisiopatologia
Il ferro è un elemento presente come gruppo eme o ferro-solfuro in numerose proteine indispensabili per la vita cellulare, ma è un potenziale agente tossico pro-ossidante se presente in forma libera. Per tale ragione gli organismi viventi hanno sviluppato meccanismi di controllo della captazione/assorbimento, utilizzo e deposito del ferro sia a livello cellulare che sistemico (3,4). Il peptide epatico epcidina è il principale regolatore sistemico dell’omeostasi del ferro. Infatti, legando e bloccando la ferroportina, l’unico noto esportatore cellulare di ferro, modula in senso inibitorio l’assorbimento intestinale del ferro e il suo rilascio dal macrofago e dall’epatocita. HFE, TFR2, e HJV, posti sulla membrana della cellula epatica agiscono come sensori del ferro circolante e del ferro di deposito e sono regolatori positivi del gene HAMP che codifica per epcidina.
 Tutte le forme di HC condividono un fenotipo comune che è il risultato della ridotta o assente sintesi di epcidina (HFE-, HJV-, HAMP-, TFR2-HC) o dalla resistenza della ferroportina all’azione di epcidina nella forma SLC40A1-correlata. Ciò determina un aumento dell’assorbimento intestinale del ferro dietetico e del suo rilascio dal macrofago da cui consegue l’aumento dei livelli di sideremia e della saturazione della transferrina (TSAT), la comparsa di ferro non legato alla transferrina (NTBI) e del suo componente LPI (ferro labile plasmatico) nel sangue. Entrambi possono penetrare le cellule in modo sregolato e alterare il delicato equilibrio del ferro intracellulare, attivare i processi ossidativi e contribuire al danno cellulare (5). I principali bersagli della tossicità da ferro sono fegato, pancreas endocrino, cuore e adenoipofisi e le principali complicanze cliniche dell’HC conclamata sono la cirrosi epatica ed eventualmente l’epatocarcinoma, il diabete, l’ipogonadismo ipofisario, lo scompenso cardiaco e l’artropatia (1,2). La gravità delle manifestazioni cliniche dipende dalla quantità e dalla rapidità dell’accumulo di ferro e dalla capacità di cellule e tessuti nel far fronte al danno ossidativo ferro-dipendente. Penetranza ed espressione dell’HC variano in relazione al gene causale, alla severità della mutazione e alla presenza di fattori addizionali genetici o acquisiti (1,2). È importante distinguere tra penetranza biochimica (aumento della TSAT e della ferritina) e clinica (evidenza del sovraccarico di ferro e delle sue complicanze). Maggiore è la repressione della sintesi di epcidina, maggiore la severità della patologia, che è massima nelle forme giovanili dovute a mutazioni dei geni HAMP e HJV (si veda rivista MR 2022;2:10-13). L’HFE-HC è caratterizzata da una marcata variabilità fenotipica, mentre la forma TFR2-correlata è considerata una forma di gravità intermedia (2).
Tutte le forme di HC condividono un fenotipo comune che è il risultato della ridotta o assente sintesi di epcidina (HFE-, HJV-, HAMP-, TFR2-HC) o dalla resistenza della ferroportina all’azione di epcidina nella forma SLC40A1-correlata. Ciò determina un aumento dell’assorbimento intestinale del ferro dietetico e del suo rilascio dal macrofago da cui consegue l’aumento dei livelli di sideremia e della saturazione della transferrina (TSAT), la comparsa di ferro non legato alla transferrina (NTBI) e del suo componente LPI (ferro labile plasmatico) nel sangue. Entrambi possono penetrare le cellule in modo sregolato e alterare il delicato equilibrio del ferro intracellulare, attivare i processi ossidativi e contribuire al danno cellulare (5). I principali bersagli della tossicità da ferro sono fegato, pancreas endocrino, cuore e adenoipofisi e le principali complicanze cliniche dell’HC conclamata sono la cirrosi epatica ed eventualmente l’epatocarcinoma, il diabete, l’ipogonadismo ipofisario, lo scompenso cardiaco e l’artropatia (1,2). La gravità delle manifestazioni cliniche dipende dalla quantità e dalla rapidità dell’accumulo di ferro e dalla capacità di cellule e tessuti nel far fronte al danno ossidativo ferro-dipendente. Penetranza ed espressione dell’HC variano in relazione al gene causale, alla severità della mutazione e alla presenza di fattori addizionali genetici o acquisiti (1,2). È importante distinguere tra penetranza biochimica (aumento della TSAT e della ferritina) e clinica (evidenza del sovraccarico di ferro e delle sue complicanze). Maggiore è la repressione della sintesi di epcidina, maggiore la severità della patologia, che è massima nelle forme giovanili dovute a mutazioni dei geni HAMP e HJV (si veda rivista MR 2022;2:10-13). L’HFE-HC è caratterizzata da una marcata variabilità fenotipica, mentre la forma TFR2-correlata è considerata una forma di gravità intermedia (2).
Emocromatosi HFE-correlata
 L’HFE-HC è una malattia ereditaria autosomico recessiva estremamente rara nelle popolazioni asiatiche e africane, rara nei sudamericani, e relativamente frequente nelle popolazioni caucasiche nord-europee (0,44%) con un picco in Irlanda (1,2%) (6). In Italia, la prevalenza è circa lo 0,2% nelle aree del nord subalpino e minore allo 0,05% nel centro-sud. Questa variabilità è legata all’origine della mutazione più comune (p.Cys282Tyr) avvenuta attorno al 4000 A.C. nell’Europa del centro-nord e alle migrazioni di queste popolazioni (7). Il genotipo più comune è lo stato di omozigosi per la variante p.Cys282Tyr presente nel 90% circa di tutti i soggetti con HFE-HC. La variante p.His63Asp del gene HFE è un polimorfismo comune la cui frequenza allelica è del 13,6% in Europa e i doppi eterozigoti p.Cys282Tyr/p.His63Asp e gli omozigoti p.His63Asp sono riportati rispettivamente nel 5% e 2% circa dei pazienti con HC. Gli omozigoti p.Cys282Tyr presentano un’elevata penetranza biochimica (elevata TSAT e ferritina), ma una penetranza clinica incompleta e un’espressione variabile. Questo significa che solo una parte dei soggetti omozigoti p.Cys282Tyr (il 30-40% dei maschi e meno del 10% delle donne) svilupperanno le complicanze cliniche del sovraccarico di ferro. La penetranza biochimica e clinica nei doppi eterozigoti p.Cys282Tyr/p.His63Asp e omozigoti p.His63Asp è molto bassa e questi genotipi non sono in grado di determinare né un sovraccarico di ferro significativo né le relative complicanze cliniche. Altri profili genetici possono causare l’HFE-HC, specialmente i doppi eterozigoti per la mutazione p.Cys282Tyr e mutazioni rare di HFE, casi singoli di omozigosi per mutazioni rare e la delezione completa del gene riscontrata esclusivamente in pazienti originari della Sardegna (Tab. 1). Lo stile di vita, il genere, la coesistenza del trait beta-talassemico e il background genetico possono modulare il fenotipo clinico (2).
L’HFE-HC è una malattia ereditaria autosomico recessiva estremamente rara nelle popolazioni asiatiche e africane, rara nei sudamericani, e relativamente frequente nelle popolazioni caucasiche nord-europee (0,44%) con un picco in Irlanda (1,2%) (6). In Italia, la prevalenza è circa lo 0,2% nelle aree del nord subalpino e minore allo 0,05% nel centro-sud. Questa variabilità è legata all’origine della mutazione più comune (p.Cys282Tyr) avvenuta attorno al 4000 A.C. nell’Europa del centro-nord e alle migrazioni di queste popolazioni (7). Il genotipo più comune è lo stato di omozigosi per la variante p.Cys282Tyr presente nel 90% circa di tutti i soggetti con HFE-HC. La variante p.His63Asp del gene HFE è un polimorfismo comune la cui frequenza allelica è del 13,6% in Europa e i doppi eterozigoti p.Cys282Tyr/p.His63Asp e gli omozigoti p.His63Asp sono riportati rispettivamente nel 5% e 2% circa dei pazienti con HC. Gli omozigoti p.Cys282Tyr presentano un’elevata penetranza biochimica (elevata TSAT e ferritina), ma una penetranza clinica incompleta e un’espressione variabile. Questo significa che solo una parte dei soggetti omozigoti p.Cys282Tyr (il 30-40% dei maschi e meno del 10% delle donne) svilupperanno le complicanze cliniche del sovraccarico di ferro. La penetranza biochimica e clinica nei doppi eterozigoti p.Cys282Tyr/p.His63Asp e omozigoti p.His63Asp è molto bassa e questi genotipi non sono in grado di determinare né un sovraccarico di ferro significativo né le relative complicanze cliniche. Altri profili genetici possono causare l’HFE-HC, specialmente i doppi eterozigoti per la mutazione p.Cys282Tyr e mutazioni rare di HFE, casi singoli di omozigosi per mutazioni rare e la delezione completa del gene riscontrata esclusivamente in pazienti originari della Sardegna (Tab. 1). Lo stile di vita, il genere, la coesistenza del trait beta-talassemico e il background genetico possono modulare il fenotipo clinico (2).
Diagnosi
 La strategia diagnostica si basa sui punti seguenti:
La strategia diagnostica si basa sui punti seguenti:
- Il sospetto clinico e biochimico. Alcuni segni e sintomi (astenia inspiegata, artralgie, riduzione della libido, e lieve aumento delle transaminasi) possono suggerire l’HC anche se nessuno di essi è specifico per la malattia. Altri, come disfunzione erettile, amenorrea primaria o secondaria, diabete, epatomegalia, cardiopatia (aritmie e scompenso) sono espressione di forme severe e avanzate della malattia. In molti pazienti la malattia è asintomatica e va cercata attraverso test di primo livello (sideremia, transferrina, e ferritina sierica). Una TSAT >45% è un segno distintivo seppur non patognomonico di HC e precede l’incremento della ferritina i cui valori massimi di normalità variano in relazione all’età e al sesso e nell’adulto sono 350-400 μg/L nell’uomo e 160-250 μg/L nella donna prima o dopo la menopausa.
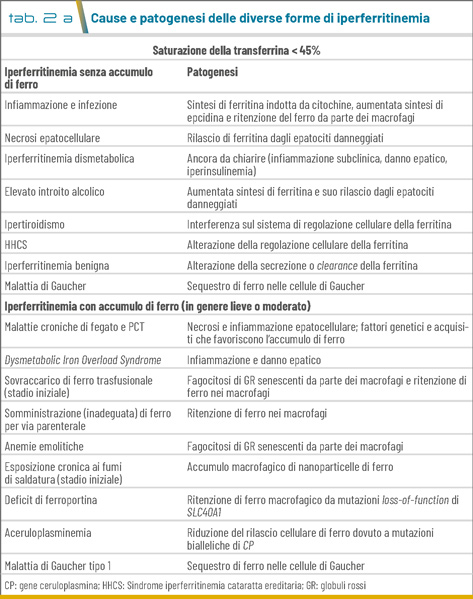
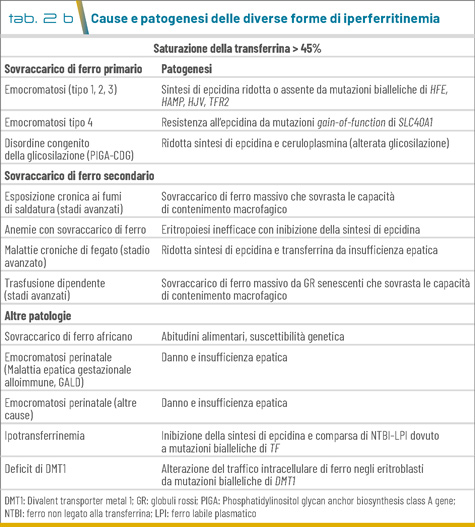
- L’esclusione di altre cause di sovraccarico di ferro o iperferritinemia. Oltre alle altre forme di HC, diverse cause acquisite o ereditarie possono determinare incrementi della ferritina, TSAT e sovraccarico di ferro, così come iperferritinemia isolata (TSAT normale) con o senza sovraccarico di ferro (Tab. 2 a e b) (1,2). È indispensabile considerare correttamente queste forme di iperferritinemia per evitare richieste non giustificate di consulenze, test genetici e diagnosi errate (8).
- La conferma del sovraccarico di ferro. La ferritina è un indice del ferro di deposito, ma è influenzata da stati infiammatori, danno epatico e problematiche metabolico-dietetiche. Se necessario, è possibile quantificare il ferro epatico mediante risonanza magnetica (RM). La biopsia epatica è oggi limitata ai casi con ferritina >1000 μg/L che definisce la soglia di rischio per il danno epatico (1). L’elastografia può essere un’alternativa non invasiva per la stima della fibrosi epatica.
- Il test genetico molecolare. Ha due obiettivi: confermare la diagnosi nel probando ed identificare il rischio genetico nei parenti. Il test genetico dovrebbe essere eseguito solo nei soggetti che presentano un’elevata TSAT e ferritina sierica, confermata in due controlli e non spiegata da altre cause (1,2,9). Nel caso di elevata TSAT ma ferritina normale, un controllo esami dopo 1-3 anni può essere una ragionevole opzione. Le tabelle 2 a e b riportano frequenza e penetranza dei diversi genotipi HFE. I casi con fenotipo emocromatosico e genotipo HFE coerente hanno diritto all’esenzione per patologia rara (RCG100) relativamente alle indagini e terapie correlate.
Screening familiare
Una volta identificato il probando, i familiari vanno invitati ad eseguire le analisi degli indici del ferro e il test genetico insieme o in sequenza. Poiché l’HFE-HC è una malattia recessiva, i soggetti più a rischio sono i fratelli/sorelle del probando (25% di probabilità di essere affetti o sani, 50% di essere portatori), mentre i figli sono portatori obbligati a meno che l’altro genitore non sia un portatore della mutazione (coppia omo-eterozigote), condizione in cui il rischio di ereditare la malattia da parte dei figli è del 50%. Come indicato dalle linee guida internazionali (9) non c’è indicazione ad eseguire il test genetico nei minori perché la malattia si manifesta in età adulta e le indagini possono tranquillamente essere posposte dopo i 18 anni, quando il ragazzo potrà esprimere il suo consenso al test genetico. Eccezione a questa regola è la presenza di precoci e significative alterazioni degli indici del ferro, improbabili nella HFE-HC, ma possibili nelle forme giovanili.
Terapia
La salassoterapia è la terapia di elezione una volta accertata la presenza di un sovraccarico di ferro. Essa consiste in una fase di induzione per rimuovere il ferro in eccesso che prevede un salasso il cui volume varia in funzione del peso (375-450 mL). La frequenza va definita in base alla severità del sovraccarico di ferro (ogni 1-4 settimane) e monitorata con emocromo, ferritina e TSAT ogni 4-6 salassi (10). Una volta raggiunta la ferrodeplezione (ferritina attorno a 50 µg/L), va impostata una terapia di mantenimento con un salasso ogni 2-6 mesi; in questa fase è possibile indirizzare il paziente alla donazione (DM 2/11/2015 – Allegato IV, sez. 1.7). Alternative alla salassoterapia quali eritrocitoaferesi e farmaci ferrochelanti vanno valutati nel singolo caso (si veda PDTA malattierare.marionegri.it).
Conclusioni
Il test genetico HFE è uno dei test genetici più frequentemente prescritti, ma esistono ancora problemi relativi ai suoi corretti utilizzo ed interpretazione che si traducono in richieste inutili, diagnosi, certificazioni di esenzione e terapie errate. È importante che i medici di medicina generale e gli specialisti si riferiscano alle linee guida internazionali (9) ed ai PDTA regionali. Il test genetico va proposto solo nel contesto di alterazioni biochimiche coerenti con l’ipotesi diagnostica. I casi dubbi vanno indirizzati ai centri di riferimento per gli approfondimenti diagnostici e la ricerca di mutazioni rare HFE e/o non-HFE, mediante tecniche di sequenziamento Sanger o di nuova generazione (NGS).
Bibliografia
- Brissot P, Pietrangelo A, Adams PC, et al. Haemochromatosis. Nat Rev Dis Primers. 2018;4:18016.
- Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Transl Gastroenterol Hepatol. 2020;5:25.
- Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28:197-213.
- Katsarou A, Pantopoulos K. Basics and principles of cellular and systemic iron homeostasis. Mol Aspects Med. 2020;75:100866.
- Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65-87.
- Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. Am J Epidemiol. 2001;154:193-206.
- Distante S, Robson KJ, Graham-Campbell J, et al. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum Genet. 2004;115:269-279.
- Piperno A, Pelucchi S, Mariani R. Hereditary Hyperferritinemia. Int J Mol Sci. 2023;24(3):2560.
- easloffice@easloffice.eu EAftSotLEa, Liver EAftSot. EASL Clinical Practice Guidelines on haemochromatosis. J Hepatol. 2022;77:479-502.
- Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317-325.