




la Newsletter
La fibrosi cistica | La fibrosi cistica (FC) è una malattia...
La fibrosi cistica (FC) è una malattia genetica a trasmissione...
La fibrosi cistica (FC) è una malattia genetica a trasmissione autosomica recessiva e coinvolgimento multisistemico, che colpisce principalmente l’apparato respiratorio e intestinale (1-3).
L’incidenza di FC varia nelle diverse etnie (1). In Italia, i dati riportano un’incidenza compresa tra 1/2.730 e 1/3.170 nati (4), per un totale di 5.362 pazienti (5).
Il gene responsabile della malattia, scoperto nel 1989 (6), si trova sul braccio lungo del cromosoma 7 (7q31.2), si estende per oltre 250.000 basi e contiene 27 esoni. La funzione principale della proteina codificata, chiamata Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), riguarda il trasporto transmembrana del cloro. Ad oggi sono state individuate più di 2.000 mutazioni del gene CFTR (7).
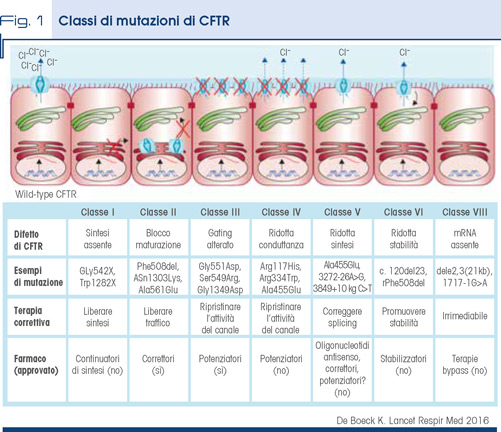 Le mutazioni del CFTR sono state classificate in base al difetto funzionale che comportano a livello della proteina CFTR in sette classi (Fig. 1).
Le mutazioni del CFTR sono state classificate in base al difetto funzionale che comportano a livello della proteina CFTR in sette classi (Fig. 1).
Le mutazioni di classe I, II e III sono comunemente associate a fenotipo più severo, con incidenza maggiore di ileo da meconio, insufficienza pancreatica, malnutrizione, deterioramento precoce e severo della funzione polmonare, malattia epatica grave. Le classi IV e V sono invece associate a malattia polmonare lieve, a funzionalità pancreatica conservata e ad una aspettativa di vita maggiore, e tendono a risultare fenotipicamente dominanti nel caso si presentino in associazione alle mutazioni di classe I-III (9,10).
CFTR è una proteina della famiglia delle ATP Binding Cassette che utilizza ATP cellulare per regolare il flusso del cloro e di altri anioni attraverso la membrana.
CFTR è espresso nelle cellule epiteliali delle vie aeree, del digerente, delle ghiandole sudoripare, e del genito-urinario. Si trova a livelli inferiori anche in cellule non epiteliali, in tessuti non coinvolti dalla malattia, come cornea, endotelio vascolare, dove forse l’espressione di altri geni è in grado di compensare la perdita del CFTR (11). Nei dotti sudoripari CFTR guida il riassorbimento di sali, mentre nell’epitelio intestinale, nei dotti pancreatici e nelle vie respiratorie, è promotore della secrezione di ione cloro e bicarbonato.
Manifestazioni cliniche
Gli aspetti clinici correlano con l’attività residua di CFTR. I pazienti con meno dell’1% di attività hanno malattia polmonare e insufficienza pancreatica; la funzione pancreatica è risparmiata con il 5% di attività residua; individui con più del 10% di attività presentano assenza bilaterale congenita dei deferenti (CBAVD) o pancreatite cronica idiopatica (1).
Le manifestazioni cliniche della malattia sono caratterizzate dalla presenza di secrezioni esocrine mucose dense che portano, nel caso dell’apparato respiratorio, ad una malattia polmonare cronica ostruttiva con evoluzione verso l’insufficienza respiratoria (12).
Alcuni tratti della malattia hanno una bassa penetranza (ileo da meconio, malattia epatica, diabete), altri hanno una variabile espressività (malattia polmonare), con un ampio spettro di gravità.
Le modalità di comparsa, l’entità dei sintomi e il decorso sono molto variabili.
Apparato respiratorio
I pazienti possono restare asintomatici a lungo o avere intermittenti e prolungate infezioni respiratorie (1,13). La pneumopatia evolve con fasi acute o subacute, dette di esacerbazione polmonare, le quali possono richiedere l’ospedalizzazione. L’insufficienza respiratoria, il cor polmonare sono l’evoluzione naturale della malattia. Il quadro clinico viene anche influenzato dall’infezione e/o colonizzazione delle vie respiratorie da parte di batteri in prevalenza gram negativi (P. aeruginosa, B. cepacia, A. xylosoxidans, S. malthophilia).La valutazione periodica dei pazienti FC include i test di funzionalità polmonare, spirometria, pletismografia, DLCO, N2-Multiple Breath Washout, colture per i patogeni associati a FC nelle vie respiratorie e HRCT.
Apparato gastrointestinale
Nell’intestino la disfunzione di CFTR comporta una disidratazione del lume intestinale con possibili manifestazioni occlusive sia a carico del tenue che del crasso (14,15). A livello della colecisti e vie biliari l’alterata secrezione di sali e acqua può condurre a colecistiti e colelitiasi. La disfunzione epatica si ritrova nel 30% dei pazienti, ma la cirrosi biliare con insufficienza epatica è sintomatica solo nel 2-3% con ittero, ascite, ematemesi da varici, ipersplenismo. Circa il 90% dei pazienti presenta insufficienza pancreatica esocrina, causa di malassorbimento, steatorrea, difetto accrescitivo ma facilmente correggibile con l’estratto pancreatico di sintesi; il diabete correlato a FC (CFRD), considerato un’entità clinica distinta dal diabete tipo 1 e 2, ha invece una prevalenza molto inferiore, 19% negli adolescenti e 40-50% negli adulti, rispettivamente (16).
Apparato riproduttivo
Pubertà ritardata: comune sia nel maschio che nella femmina con FC. Azoospermia nel 95%, amenorrea secondaria, cervicite e accumulo di muco cervicale denso nelle femmine con conseguente riduzione della fertilità. La gravidanza in FC è descritta però con sempre più frequenza ed il FEV1% pre-gravidanza sembra essere il predittore di outcome più utile (17,18).
Ghiandole sudoripare
L’eccessiva perdita di sali può predisporre i bambini a squilibri elettrolitici, specialmente durante episodi di gastroenterite o in presenza di temperature elevate, e provocare disidratazioni anche gravi con alcalosi ipocloremica (1).
Diagnosi
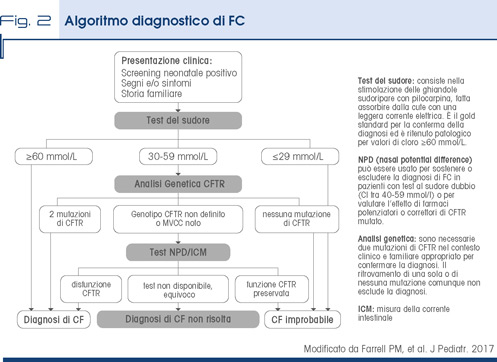 Algoritmo diagnostico
Algoritmo diagnostico
La diagnosi di FC si basa sulla combinazione di manifestazioni cliniche con il rilievo di anormalità di CFTR ad uno dei tre test diagnostici validati. Questi comprendono il test del sudore (con cloro e sodio superiori ai limiti di norma), l’analisi genetica che si considera positiva quando identifica due mutazioni che causano la malattia, lo studio della differenza di potenziale elettrico transepiteliale nelle mucose respiratorie o intestinali (Fig. 2).
Screening neonatale
La metodologia è varia e include il test del tripsinogeno immunoreattivo (IRT) combinato con l’analisi del DNA, IRT ripetuto (double IRT testing) e il test della proteina associata alla pancreatite (PAP). La conferma definitiva o l'esclusione della diagnosi viene comunque affidata al test del sudore, con richiamo del neonato a 20-30 giorni di vita. Questo screening raggiunge una sensibilità del 90%. È una metodica ormai diffusa in quasi tutte le regioni d’Italia. Lo screening consente la presa in carico da parte di centri specializzati più precocemente che con la diagnosi fatta per sintomi (19).
Screening del portatore
Lo screening al portatore è stato storicamente riservato nel caso di anamnesi familiare positiva per CF, o nel caso di partner di soggetti affetti da CF. In alcune regioni è stato proposto uno screening di popolazione, il cui vantaggio principale è di individuare adulti eterozigoti e consentire in tal modo scelte riproduttive informate, in seguito a counseling genetico (20).
Terapia
Capisaldi del trattamento dopo la diagnosi sono l’educazione del paziente e della famiglia, e un follow-up intensivo con controlli ogni 2-3 mesi. L’approccio è multidisciplinare e finalizzato a promuovere la clearance delle secrezioni, gestire le infezioni polmonari, mantenere un adeguato stato nutrizionale, supplire alle carenze nutritive legate al malassorbimento, prevenire l’ostruzione intestinale, mantenere l’omeostasi glucidica, per conservare una condizione clinica stabile per periodi prolungati.
Trattamento della malattia polmonare
Sono fondamentali il controllo delle infezioni tramite l’analisi colturale dell’escreato bronchiale o dell’aspirato faringeo, il monitoraggio della funzionalità respiratoria e le vaccinazioni, soprattutto verso i patogeni che coinvolgono l’apparato respiratorio. Il trattamento si avvale di terapie non farmacologiche, quali la fisioterapia respiratoria e l’esercizio fisico, e farmacologiche quali antibiotici inalatori o sistemici, antinfiammatori steroidei inalatori e sistemici, e non steroidei sistemici, broncodilatatori inalatori a breve e lunga durata d’azione, e mucolitici.
L’ossigeno-supplementazione si rende necessaria negli stadi avanzati di malattia, inizialmente nelle ore del riposo notturno e durante lo sforzo, poi con il progredire della patologia, anche a riposo.
Il supporto ventilatorio con CPAP o BiPAP può essere un valido ausilio per la fisioterapia, ma può diventare un provvedimento necessario ai fini della ventilazione nelle fasi avanzate di malattia e insufficienza respiratoria.
Il trapianto polmonare è l’ultimo step terapeutico e risulta vantaggioso quando la sopravvivenza stimata a 5 anni senza trapianto risulti inferiore al 30%. Indicazioni al trapianto sono: insufficienza respiratoria cronica con ossigenodipendenza, FEV1<30% del predetto, rapido declino del FEV1 con particolare attenzione da rivolgere ai pazienti giovani, o con emottisi, pneumotorace refrattario o ricorrente e aumento della frequenza delle esacerbazioni.
Recentemente sono entrati a far parte del protocollo terapeutico FC nuovi farmaci che modulano la funzione del CFTR.
I farmaci potenziatori, di cui ivacaftor è stato il primo approvato dalla FDA nel 2012, sono molecole che aumentano la probabilità di apertura del canale CFTR, migliorandone la conduttanza, fino al 35-40% dell’attività normale (21,22); sono pertanto indicati nelle mutazioni gating, in cui il CFTR è presente sulla membrana cellulare ma presenta un difetto di apertura o una conduttanza ridotta.
L’utilizzo di ivacaftor nelle mutazioni gating come G551D ha dimostrato prolungato beneficio in termini di test del sudore, FEV1, BMI e frequenza di esacerbazioni.
Sono successivamente state sviluppate molecole che agiscono da correttori, con un’attività rescue sul ripiegamento e sul trasporto del CFTR, che risultando più stabile, viene trasportato sulla membrana plasmatica. La combinazione di ivacaftor con il correttore lumacaftor, approvata nel 2015 dalla FDA per soggetti omozigoti per F508del, ha mostrato vantaggi a breve e lungo termine sulla funzionalità polmonare soprattutto come riduzione del declino polmonare annuo atteso, e sul BMI (23,24).
La combinazione di ivacaftor con un altro correttore, tezacaftor, è stata approvata dalla FDA nel 2018 per pazienti maggiori a 12 anni con F508del in omozigosi, o in eterozigosi con funzione residua, con beneficio sulla funzione respiratoria e sulla qualità di vita (25).
La triplice combinazione di elexcaftor/ tezacaftor/ivacaftor è l’ultimo dei farmaci approvati dalla FDA, nel 2019, e dall’EMA ad agosto 2020, per i pazienti con F508del in omozigosi, o in eterozigosi con funzione minima, dopo i promettenti risultati in termini di aumento del FEV1, riduzione di cloro al test del sudore, e miglioramento dei punteggi di qualità di vita, con un profilo di sicurezza accettabile (26).
Prognosi
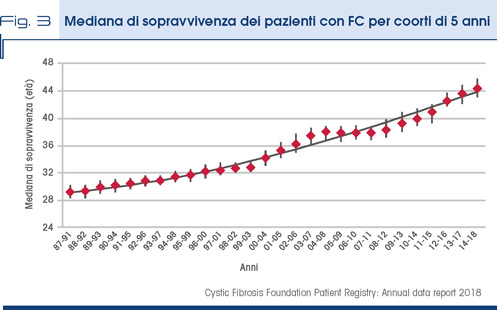 Grazie a queste innovazioni terapeutiche la sopravvivenza dei pazienti FC è molto migliorata negli ultimi decenni. Gli ultimi dati della CF Foundation americana descrivono una mediana di sopravvivenza per i nati nel 2018 di 47,4 anni (IC 95% 44.2–50.3 anni) destinata ulteriormente ad aumentare grazie alle nuove terapie (Fig. 3).
Grazie a queste innovazioni terapeutiche la sopravvivenza dei pazienti FC è molto migliorata negli ultimi decenni. Gli ultimi dati della CF Foundation americana descrivono una mediana di sopravvivenza per i nati nel 2018 di 47,4 anni (IC 95% 44.2–50.3 anni) destinata ulteriormente ad aumentare grazie alle nuove terapie (Fig. 3).
Bibliografia
- Egan ME, et al. Cystic fibrosis. In: Nelson TEXTBOOK of PEDIATRICS 20th EDITION. Elsevier Ltd; 2016:3106-3123.
- Elborn JS. Cystic Fibrosis. Lancet. 2016:388:2519-2531.
- Braggion C, Piacentini G, Boner AL. Fibrosi cistica. In: Edra, ed. Rugarli. Medicina Interna Sistematica. Vol 1; 2017:482-490.
- Castellani C, Massie J, Sontag M, et al. Newborn screening for cystic fibrosis. Lancet Respir Med. 2016;4(8):653-661.
- Giordani B, Amato A, Majo F, et al. Gruppo di lavoro RIFC. Italian Cystic Fibrosis Registry (ICFR). Report 2015-2016. Epidemiol Prev. 2019 Jul-Aug;43(4S1):1-36.
- Kerem B, Rommens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science (80- ). 1989;121(August):245:1073-80
- Cystic Fibrosis Genetic Consortium. Cystic Fibrosis Genetic Data Base. URL: htpp://www.genet. sickkids.on.ca/cftr.
- De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662-674.
- Castellani C, Cuppens H, Macek M, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros. 2008;7(3):179-196.
- Fajac I, De Boeck K. New horizons for cystic fibrosis treatment. Pharmacol Ther. 2017;170:205-211.
- Trezise AEO: Exquisite and Multilevel Regulation of CFTR Expression. In: Cystic Fibrosis in the 21st Century. Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A eds. Prog Respir Res. Basel, Karger, 2006, vol 34, pp 11-20.
- Boucher RC. Airway Surface Dehydration in Cystic Fibrosis: Pathogenesis and Therapy. Annu Rev Med. 2007;58(1):157-170.
- Katkin J. Cystic fibrosis: Clinical manifestations and diagnosis. Up to Date 2019.
- Gorter RR, Karimi A, Sleeboom C, et al. Clinical and genetic characteristics of meconium ileus in newborns with and without cystic fibrosis. J Pediatr Gastroenterol Nutr. 2010;50(5):569-572.
- Dray X, Bienvenu T. Distal Intestinal Obstruction Syndrome in Adults with Cystic fibrosis. Clin Gastroenterol Hepatol. 2004;15(04):175-182.
- Kelly A, Moran A. Update on cystic fibrosis-related diabetes. J Cyst Fibros. 2013;12(4):318-331.
- Dupuis A, Corey M, Tullis DE. Pregnancy in Cystic Fibrosis * Fetal and Maternal Outcome. Chest. 2000;118(1):85-91.
- Thorpe-Beston JG: Fertility, Contraception, Incontinence and Pregnancy. In: Cystic Fibrosis in the 21st Century. Bush A, Alton EWFW, Davies JC, Griesenbach U, Jaffe A eds. Prog Respir Res. Basel, Karger, 2006, vol 34, pp 264-269.
- Farrell PM, White TB, Ren CL, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181:S4-S15.e1.
- Castellani C, Massie J. Newborn screening and carrier screening for cystic fibrosis: alternative or complementary? Eur Respir J. 2014;43(1):20-23.
- Harutyunyan M, Huang Y, Mun K-S, et al. Personalized medicine in CF: from modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am J Physiol Cell Mol Physiol. 2018;314(4): L529-L543.
- Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir Med. 2013;1(2):158-163.
- Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783- 1784.
- Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5(2):107-118.
- Donaldson SH, Pilewski JM, Griese M et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-224.
- Keating D, Marigowda G, Burr L, et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med. 2018;379(17):1612-1620.
- Cystic Fibrosis Foundation Patient Registry: Annual data report 2018. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient- Registry-Annual-Data-Report.pdf