




la Newsletter

Silvia Bilia, Antonio Tavoni
U.O. Immunologia Clinica - Dipartimento di Medicina Clinica e Sperimentale, Università di Pisa
La malattia da IgG4 | La IgG4-RD è una rara malattia...
La IgG4-RD è una rara malattia fibro-infiammatoria caratterizzata...
La malattia da IgG4 (IgG4-Related disease, IgG4-RD) è una rara malattia fibro-infiammatoria caratterizzata dallo sviluppo di masse pseudo-tumorali in vari distretti, costituite da infiltrati di plasmacellule IgG4-positive e fibrosi storiforme, spesso accompagnate da aumento delle IgG4 sieriche.
Descritta per la prima volta come entità clinica nel 2003, sono confluite nella sua definizione varie condizioni prima distinte, tra cui pancreatite autoimmune di tipo 1, malattia di Mikulicz, tiroidite di Riedel, fibrosi retroperitoneale. L’incidenza e la prevalenza rimangono sottostimate, da 0.26 a 1.08 casi per 100.000 abitanti l’anno. L’andamento cronico e la buona risposta alla terapia immunosoppressiva hanno suggerito una patogenesi autoimmune, ancora in gran parte ignota.
Aspetti fisiopatologici
 Dal punto di vista fisiopatologico si può distinguere un andamento “bifasico”: un’iniziale fase infiammatoria sostenuta da cellule B e T (circolanti e tissutali) dirette contro uno o più specifici autoantigeni, lascia poi il posto ad esiti fibrotici con possibile danno di un organo affetto. La natura dell’antigene e il motivo per cui vi siano specifici tessuti bersaglio sono in fase di studio.
Dal punto di vista fisiopatologico si può distinguere un andamento “bifasico”: un’iniziale fase infiammatoria sostenuta da cellule B e T (circolanti e tissutali) dirette contro uno o più specifici autoantigeni, lascia poi il posto ad esiti fibrotici con possibile danno di un organo affetto. La natura dell’antigene e il motivo per cui vi siano specifici tessuti bersaglio sono in fase di studio.
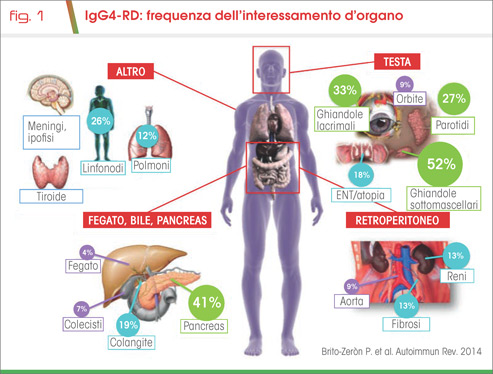
La malattia ha un andamento remittente-recidivante, con ampia variabilità degli organi colpiti, interessati in modo sincrono o in diverse fasi (Fig. 1) (Tab. 1).
La diagnosi prevede un corretto inquadramento clinico-laboratoristico che richiede spesso una biopsia di conferma (dove eseguibile). La diagnosi differenziale è con patologie neoplastiche, infettive e connettiviti/vasculiti.
Manifestazioni cliniche
 Il pancreas è l’organo più frequentemente interessato: si presenta radiologicamente ispessito, con edema diffuso intorno alla ghiandola (aspetto “sausage-like”); clinicamente è possibile ittero, prurito, dolore addominale, fino a insufficienza esocrina ed endocrina ghiandolare. Nel 60-80% dei casi la pancreatite si associa a colangite sclerosante IgG4-relata. Possibile inoltre una colecistite IgG4-relata, spesso di riscontro accidentale (da imaging o per colecistectomia nel sospetto di carcinoma).
Il pancreas è l’organo più frequentemente interessato: si presenta radiologicamente ispessito, con edema diffuso intorno alla ghiandola (aspetto “sausage-like”); clinicamente è possibile ittero, prurito, dolore addominale, fino a insufficienza esocrina ed endocrina ghiandolare. Nel 60-80% dei casi la pancreatite si associa a colangite sclerosante IgG4-relata. Possibile inoltre una colecistite IgG4-relata, spesso di riscontro accidentale (da imaging o per colecistectomia nel sospetto di carcinoma).
Le ghiandole salivari (parotidi, sottomandibolari e sottolinguali) sono spesso interessate. Più frequentemente sono coinvolte le sottomandibolari con tumefazione, scialoadeniti e xerostomia. L’interessamento delle ghiandole lacrimali è spesso associato a quello delle salivari (10-50% dei casi, cosiddetta “sindrome di Mikulicz”): clinicamente è presente tumefazione indolore delle ghiandole con o senza proptosi, o pseudotumor che interessa i tessuti periorbitari in genere senza compromissione del visus. Il coinvolgimento orbitario da IgG4-RD costituisce 1/5 delle patologie infiammatorie e tumorali interessanti l’orbita; necessario escludere un morbo di Graves, sarcoidosi, granulomatosi con poliangioite, linfomi ed infezioni.
La fibrosi retroperitoneale è frequente, si presenta con dolore addominale o lombare o è di riscontro occasionale all’imaging, l’infiammazione può estendersi a vie urinarie e aorta. Circa 1/5 di tutti i casi di fibrosi retroperitoneale è riconducibile alla IgG4-RD.
È possibile un coinvolgimento toracico rappresentato da aortite, linfoadenomegalia, noduli polmonari, ispessimento e/o versamento pleurico, masse mediastiniche o paravertebrali.
Il coinvolgimento renale è più spesso costituito da nefrite tubulointerstiziale ad interessamento focale, ma è possibile anche una glomerulopatia (più spesso da glomerulonefrite membranosa). Clinicamente la prima si manifesta con riduzione di funzionalità renale, blanda (non sempre presente) proteinuria/ematuria ed ipocomplementemia.
Nel 10-20% dei casi si riscontra un interessamento vascolare dei grossi vasi. La malattia predilige la tonaca avventizia dell’aorta e i tessuti periavventiziali con relativo risparmio della media. Frequente è la periaortite correlata a fibrosi retroperitoneale, da formazione di tessuto molle che va a circondare il vaso con ispessimento/enhancement di parete. Questo interessamento può essere letale potendo condurre alla formazione di aneurismi, dissezione o perforazione del vaso.
Il coinvolgimento neurologico più frequente è una pachimeningite (1/3 delle meningiti ipertrofiche è riconducibile a IgG4-RD) con clinica variabile e imaging aspecifico (ispessimento meningeo omogeneamente ipertrofico o di aspetto nodulare). Possibile inoltre un’ipofisite IgG4-relata con quadri endocrinologici secondari al danno infiammatorio di adeno e/o neuroipofisi.
Altre possibili manifestazioni di IgG4-RD sono tiroidite fibrosante, prostatite, e mesenterite sclerosante (da fibrosi della radice del mesentere) dove un debulking chirurgico costituisce un approccio sinergico alla terapia medica.
La predilezione del coinvolgimento di certi distretti in cluster ha recentemente permesso di individuare quattro “fenotipi” clinici di malattia, con caratteristiche clinico-epidemiologiche e prognostiche differenti.
Il tipo pancreato-biliare è caratterizzato prevalentemente da pancreatite autoimmune e colangite, elevate IgG4 e IgE sieriche, e buona risposta alla terapia.
Il tipo retroperitoneo e aorta si presenta per lo più con interessamento aorto-linfatico-pericardico e fibrosi retroperitoneale, bassi livelli di IgG4 sierici ed è resistente al trattamento.
Il tipo ENT limitato a testa collo (orbite, ghiandole salivari e lacrimali, tiroide, meningi, ipofisi e seni paranasali) è resistente alla terapia.
Il tipo Mikulicz e sistemico dà coinvolgimento di ghiandole salivari e sistemico (come per nefrite, pancreas, polmone o pleura), è associato ai più alti livelli sierici di IgG4 ed è responsivo alla terapia.
Diagnosi
L’istologia svolge un ruolo importante nella diagnosi. I tre principali elementi tipici sono la fibrosi storiforme, l’infiltrato linfoplasmocitico policlonale con abbondanti cellule IgG4+ e la flebite obliterante (una completa occlusione del lume di vene di piccolo e medio calibro da parte di un infiltrato infiammatorio di linfociti e plasmacellule). Seppure non patognomomico, la fibrosi storiforme è forse l’elemento più distintivo della malattia.
I pattern non si manifestano allo stesso modo in tutti gli organi colpiti. L’immunoistochimica è importante anche per quantificare il rapporto di plasmacellule IgG4+/IgG totali: un rapporto maggiore del 40% è suggestivo di diagnosi (ma valori inferiori non escludono la diagnosi a priori). Tra gli esami di laboratorio si riscontra elevazione della VES (in parte secondaria ad ipergammaglobulinemia) e PCR, eosinofilia.
Vi è un’elevazione delle IgG4 sieriche in un 70% dei pazienti: pertanto, valori elevati di IgG4 (>135 mg/dl) seppure supportano l’ipotesi diagnostica, non sono sufficientemente specifici né sensibili.
L’imaging è fondamentale: un esame funzionale come la PET-Tc è utile sia in fase diagnostica per guidare eventuali indagini istologiche, sia nel follow-up per documentare una risposta al trattamento.
 La conoscenza sempre maggiore sulla patologia ha portato al recente sviluppo di criteri classificativi (ACR/EULAR 2019) che tuttavia, nonostante una buona sensibilità e specificità (82% e 97.8%) non sono di facile applicazione nella pratica clinica. A tal proposito, i più recenti criteri diagnostici di Umehara utilizzano tre item (clinico/radiologico – sierologico – istologico) per orientare verso una diagnosi certa, probabile o possibile di IgG4-RD, risultando di più facile applicabilità routinaria (Tab.2).
La conoscenza sempre maggiore sulla patologia ha portato al recente sviluppo di criteri classificativi (ACR/EULAR 2019) che tuttavia, nonostante una buona sensibilità e specificità (82% e 97.8%) non sono di facile applicazione nella pratica clinica. A tal proposito, i più recenti criteri diagnostici di Umehara utilizzano tre item (clinico/radiologico – sierologico – istologico) per orientare verso una diagnosi certa, probabile o possibile di IgG4-RD, risultando di più facile applicabilità routinaria (Tab.2).
Trattamento
La terapia di prima linea sono i corticosteroidi, di comune utilizzo con vari schemi per via orale (0.6-1 mg/kg/die di prednisone o prednisolone per 3-4 settimane con decalage nell’arco di 3-6 mesi) o endovena in caso di trattamento d’urgenza per rischio di sequele (fino a 1 grammo/die di metilprednisolone per 3 giorni consecutivi con decalage, pulse therapy).
La selezione dei pazienti che richiedono una terapia di mantenimento e le sue modalità non sono del tutto chiarite e rimangono tuttora basate sulla expert opinion.
In presenza di fattori predittori di rischio di recidiva (es. coinvolgimento multiorgano, elevato livello di IgG4 sieriche e IgE, eosinofilia periferica) è indicata una terapia di mantenimento.
Tra le terapie immunosoppressive rituximab (anticorpo monoclonale anti-CD20) rappresenta il farmaco maggiormente in grado di indurre e mantenere remissione: il dosaggio di 1 grammo endovena in 2 infusioni (0-15 giorni) seguito da infusioni di mantenimento ogni sei mesi circa rappresenta lo schema più utilizzato nella pratica clinica.
Tra gli altri DMARDs la ciclofosfamide rappresenta una valida alternativa al rituximab. Micofenolato, abatacept, methotrexate, anti-TNF hanno mostrato minor efficacia e sono pertanto meno utilizzati; ne è possibile un utilizzo in combinazione nel mantenimento della remissione (es. per il methotrexate).
La chirurgia è consigliata nei pazienti con sintomi ostruttivi, infiltrativi o compressivi. La sempre più ampia conoscenza dei meccanismi patogenetici sta portando allo sviluppo di nuove molecole target (come anti-CD19, anti-SLAMF7).
Bibliografia
- Maritati F, Peyronel F, Vaglio A.IgG4-related disease: aclinical perspective. Rheumatology (Oxford). 2020;59(3):iii123-iii131.
- Lanzillotta M, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ. 2020;369.
- MahajanVS, Mattoo H, Deshpande V, et al. IgG4-related disease. Annu Rev Pathol. 2014;9:315-47.
- Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. 2020;79(1):77-87.
- Brito-Zeròn P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203-10.
- Zhang W, Stone JH. Management of IgG4-related disease. Lancet Rheumatol. 2019;1(1):e55-e65.
- Umehara H. et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021;31(3):529-533.
- G. Costanzo, L. Puccetti, R. Capecchi, S. Bilia, I. Puxeddu, A. Tavoni, P. Migliorini. The IgG4-related disease: performance of classification and diagnostic criteria in a single-center cohort of patients. Clin Exp Rheumatol (in press)