




la Newsletter
La neurofibromatosi tipo 1: una sindrome da predisposizione ai...
La neurofibromatosi tipo 1 è una malattia genetica neurocutanea...
La neurofibromatosi tipo 1 (NF1) è una malattia genetica a trasmissione autosomica dominante, con penetranza completa entro gli 8 anni circa, a prevalente interessamento neurocutaneo associato a complicanze multisistemiche. Si tratta di una condizione panetnica e la sua incidenza è pari a 1:2.500-3.000. In circa il 50% dei casi è ereditata da un genitore, mentre nella restante porzione i soggetti affetti da NF1 sono gli unici del loro nucleo familiare (casi sporadici o de novo).
Eziologia
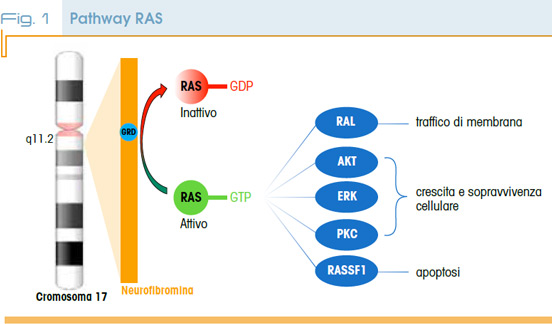 La NF1 è causata da alterazioni in eterozigosi a carico del gene onco-soppressore NF1 (17q11.2), che codifica per la proteina neurofibromina, coinvolta nella regolazione negativa del pathway RAS, implicato nella proliferazione e differenziazione cellulare (Fig. 1). Nella maggior parte dei casi si tratta di mutazioni inattivanti, che causano una loss-of-function del gene NF1, con conseguente produzione di ridotti livelli di neurofibromina; circa il 5% dei pazienti presenta invece una microdelezione a carico della regione 17q11.2 comprendente l’intero gene NF1. Ad oggi non è possibile stabilire una correlazione tra l’alterazione genetica e le eventuali manifestazioni cliniche della neurofibromatosi (correlazione genotipo-fenotipo), eccetto che per alcuni casi specifici, in particolare:
La NF1 è causata da alterazioni in eterozigosi a carico del gene onco-soppressore NF1 (17q11.2), che codifica per la proteina neurofibromina, coinvolta nella regolazione negativa del pathway RAS, implicato nella proliferazione e differenziazione cellulare (Fig. 1). Nella maggior parte dei casi si tratta di mutazioni inattivanti, che causano una loss-of-function del gene NF1, con conseguente produzione di ridotti livelli di neurofibromina; circa il 5% dei pazienti presenta invece una microdelezione a carico della regione 17q11.2 comprendente l’intero gene NF1. Ad oggi non è possibile stabilire una correlazione tra l’alterazione genetica e le eventuali manifestazioni cliniche della neurofibromatosi (correlazione genotipo-fenotipo), eccetto che per alcuni casi specifici, in particolare:
- la microdelezione 17q11.2, che è associata ad un fenotipo più severo, dato da: comparsa più precoce e numero maggiore di neurofibromi, dismorfismi facciali caratteristici, disabilità intellettiva e rischio aumentato rispetto agli altri soggetti con NF1 di sviluppare un tumore maligno della guaina dei nervi periferici [1]
- la delezione in-frame di 3bp a carico dell’esone 17, che è solitamente associata ad un fenotipo più lieve, con assenza di neurofibromi cutanei e plessiformi [2]
- la mutazione missenso c.5425C>T (p.Arg1809Cys): analogamente alla delezione sopracitata, tale alterazione sembra essere correlata ad un fenotipo lieve, caratterizzato da macchie cutanee caffelatte e feckling, in assenza di neurofibromi [3].
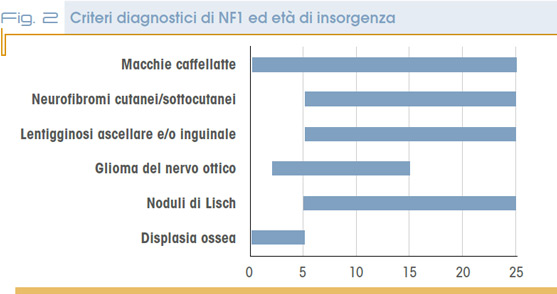
La condizione si caratterizza per la comparsa età-specifica dei segni/sintomi (cutanei e sistemici) e delle complicanze associate (Fig. 2). La diagnosi clinica si basa sulla presenza di almeno due dei criteri diagnostici definiti nel 1987 dal National Institutes of Health (NIH) Consensus Conference (Tab. 1).

Clinica
Nonostante NF1 sia conosciuta soprattutto per la componente cutanea/estetica (macchie caffèllatte, lentigginosi ascellare e inguinale, neurofibromi cutanei, sottocutanei o plessiformi), si tratta di una patologia complessa ed eterogenea, che oltre alle manifestazioni sopracitate può dare un coinvolgimento di più organi e apparati; circa un quarto dei soggetti affetti può infatti sviluppare complicanze oculistiche, ortopediche, neurologiche, internistiche, endocrinologiche e oncologiche. Rispetto alle ultime, il rischio generale di sviluppare un tumore (oltre ai neurofibromi cutanei, sottocutanei e plessiformi) nella popolazione affetta da NF1 è stimato essere superiore del 5-15% rispetto a quello della popolazione generale.
I meccanismi oncogenetici non sono ancora del tutto chiari; è noto che per il manifestarsi della maggior parte delle caratteristiche cliniche della patologia, come le macchie cutanee color caffellatte e i neurofibromi, oltre alla mutazione costituzionale (germinale) è necessario che si verifichi una seconda mutazione somatica a carico dell’allele non mutato (wild-type) del gene NF1; questa condizione (modello second-hit), tuttavia, non è sufficiente perché si sviluppi una trasformazione maligna [4]. Perché si verifichi tale evento sono infatti indispensabili ulteriori alterazioni genetiche, ad oggi non ancora del tutto determinate, tra cui mutazioni e/o modificazioni epigenetiche in altri geni che controllano la proliferazione, il differenziamento, la morte e l’integrità del patrimonio genetico cellulare, andando a costituire un processo “multistep”.
Rischio oncologico
Nonostante le complicanze della NF1 siano numerose, le neoplasie costituiscono la causa di morte più comune negli individui affetti da NF1 e provocano una riduzione dell’aspettativa di vita che può arrivare a 10-15 anni in meno rispetto alla popolazione generale [5]. Per questo motivo si è scelto, in questa sede, di approfondire tale problematica (per una review completa sulla NF1 si rimanda al recente lavoro di Gutmann et al. [6]).
Sebbene alcuni tumori NF1-associati abbiano un andamento generalmente benigno e spesso richiedano un trattamento conservativo, alcune neoplasie correlate a NF1 possono presentare un decorso severo. Il rischio oncologico aumentato riguarda più frequentemente neoplasie di origine nervosa e del tessuto connettivo, tuttavia è noto che anche altri tumori solidi possono occorrere in un individuo affetto da NF1, sia in età infantile che adulta; nella maggior parte dei casi si tratta di neoplasie molto rare nella popolazione generale e spesso l’età di insorgenza nei pazienti affetti da NF1 è ridotta rispetto ai casi sporadici.
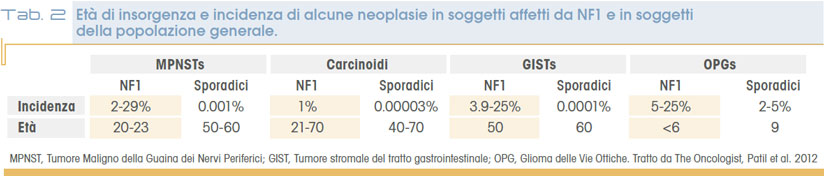
Nella tabella 2 sono riportate, a titolo di esempio, le diverse incidenze ed età di insorgenza tra popolazione NF1 e popolazione generale di quattro differenti tumori associati alla condizione in oggetto.
E’ indispensabile che il clinico di riferimento conosca tali complicanze, le loro caratteristiche cliniche e i sintomi ad esse correlati e crei con il paziente un’alleanza terapeutica, trasmettendogli le informazioni necessarie perché possa essere sensibilizzato sui sintomi di allarme; in molti di questi casi, infatti, la precocità della diagnosi è di fondamentale importanza per garantire una corretta e tempestiva presa in carico diagnostico/terapeutica, modificando anche notevolmente la prognosi della malattia.
Per una corretta presa in carico dei pazienti con NF1 sono stati definiti a livello regionale dei Percorsi Diagnostico-Terapeutici Assistenziali (PDTA), basati sulle evidenze più recenti della letteratura nazionale ed internazionale, che prevedono l’intervento di un team multidisciplinare di specialisti ed operatori sanitari (per il PDTA della Regione Lombardia si veda http://malattierare.marionegri.it/content/view/123).
Caratteristiche cliniche e approcci terapeutici
E’ bene ricordare che a seguito del sospetto clinico è opportuno indirizzare il paziente nei centri di riferimento specifici per la problematica, in modo tale che possa essere messo in atto un corretto e aggiornato percorso diagnostico e terapeutico.
Tumori del Sistema Nervoso Centrale (SNC)
I tumori del SNC si manifestano in circa il 20% dei soggetti affetti da NF1 e sono circa 5 volte più frequenti rispetto alla popolazione generale:
Glioma delle vie ottiche (OPG): si tratta di una neoplasia gliale di basso grado (astrocitoma pilocitico), che insorge solitamente entro la prima decade, con un picco tra i 4 e i 5 anni e interessa fino al 15% dei bambini con NF1 [7] [8]; sebbene possa essere colpita tutta la via ottica, nell’NF1 il tratto più frequentemente interessato è quello anteriore. I gliomi delle vie ottiche in questi casi, a differenza di quelli sporadici, presentano tipicamente un decorso indolente, con crescita lenta o nulla e talvolta con possibilità di regressione spontanea. Nella maggior parte dei casi rimangono asintomatici; solo una proporzione variabile da un terzo alla metà dei casi sviluppa sintomi, che possono essere diversi a seconda della sede interessata (proptosi, riduzione dell’acuità visiva/del campo visivo, pubertà precoce). Il trattamento di prima scelta è la chemioterapia ed è indicato solo in caso di chiara evidenza di progressione clinico-radiologica della malattia (negli altri casi si adotta un atteggiamento conservativo, con sorveglianza clinica e strumentale).
Gliomi del tronco cerebrale: costituiscono la seconda più frequente neoplasia intracranica nei pazienti con NF1; anch’essi colpiscono soprattutto in età infantile e sono localizzati prevalentemente nel midollo allungato [9]. L’istotipo più frequente è l’astrocitoma pilocitico e il decorso è generalmente benigno (a differenza dei casi sporadici), con crescita lenta e possibilità di regressioni spontanee; spesso sono sintomatici all’esordio (la cefalea costituisce il sintomo più frequente). Il trattamento solitamente è conservativo, con sorveglianza clinica e neuroradiologica; nei casi che presentano una rapida crescita o diventano sintomatici la terapia di scelta è la resezione chirurgica, quando anatomicamente possibile.
Sono descritti anche gliomi cerebellari e cerebrali (che tendono a presentare una maggiore aggressività rispetto a quelli del tratto ottico-chiasmatico e del tronco encefalico) e, più raramente, ependimomi e medulloblastomi. Nei pazienti di età superiore a 10 anni la prevalenza di gliomi di alto grado è aumentata di 50-100 volte rispetto alla popolazione generale.
Tumori extra-SNC
Tumori maligni della guaina dei nervi periferici (MPNST): si tratta di sarcomi che insorgono a partenza dalle cellule di Schwann dei nervi periferici; possono colpire qualsiasi distretto anatomico, ma si riscontrano più frequentemente nelle porzioni prossimali degli arti e nel tronco. Il rischio di sviluppare un MPNST per una persona affetta da NF1 durante l’arco della vita è pari all’8-13% [10]; la prognosi è peggiore rispetto ai casi di MPNST sporadici. Solitamente si manifestano nella II-III decade (sono rari nell’infanzia), insorgono da un neurofibroma plessiforme o sottocutaneo pre-esistente (mai da neurofibromi cutanei) ed esordiscono con dolore persistente, tipicamente notturno, e non responsivo alla terapia antidolorifica; possono inoltre essere presenti modificazioni nelle dimensioni e nella consistenza della lesione pre-esistente, comparsa di deficit neurologici inspiegati [11].
Il rischio di sviluppare un MPNST è aumentato di 3 volte in presenza di neurofibromi sottocutanei e di 10 volte in presenza di neurofibromi interni/plessiformi (talvolta la diagnosi è complessa perché originano da lesioni non rilevabili all’esame obiettivo); il rischio è aumentato anche nei soggetti che in passato hanno subito radioterapia, che presentano un’anamnesi familiare positiva per tumore, o in cui l’analisi genetica abbia messo in evidenza la presenza di una delezione dell’intero gene NF1. Il trattamento prevede la resezione completa – se possibile a seconda della localizzazione anatomica – con radioterapia adiuvante per tutte le neoplasie di grado intermedio/alto; la chemioterapia viene riservata ai casi che presentano metastasi o che non sono asportabili chirurgicamente.
Tumori stromali del tratto gastrointestinale (GIST): sono neoplasie mesenchimali che insorgono a partenza dalle cellule interstiziali di Cajal o dai loro progenitori e possono colpire qualsiasi porzione del tratto gastroenterico; tuttavia, mentre i casi sporadici insorgono prevalentemente a livello gastrico, i GIST associati ad NF1 colpiscono soprattutto il piccolo intestino e talvolta possono essere multifocali. Si stima che il rischio per una persona affetta da NF1 di sviluppare tale complicanza sia superiore di circa 45 volte rispetto a quello della popolazione generale [4].
Solitamente sono asintomatici; solo il 5% dei casi esordisce con una sintomatologia aspecifica data da dolore addominale, segni di sanguinamento gastroenterico e raramente ostruzione intestinale. La prognosi è solitamente migliore rispetto ai casi sporadici e il trattamento di scelta prevede la resezione chirurgica.
Feocromocitomi: si tratta di tumori che originano dalle cellule cromaffini del neuroectoderma a livello della midollare del surrene o dei gangli del sistema simpatico, la cui caratteristica è quella di secernere catecolamine. Colpiscono circa l’1% dei soggetti con NF1 e sono solitamente solitari e monolaterali. L’ipersecrezione di catecolamine provoca ipertensione arteriosa, che solitamente è associata a cefalea, palpitazioni, sudorazione profusa e stato d’ansia. La prognosi dei feocromocitomi associati ad NF1 è generalmente buona e il trattamento chirurgico risulta risolutivo nel 95% dei casi; la chemioterapia è riservata ai casi che presentano metastasi.
Tumore della mammella: nelle donne affette da neurofibromatosi tipo 1 è descritto un rischio aumentato di cinque volte di sviluppare tale complicanza prima dell’età di 50 anni e, in generale, un rischio di sviluppare il tumore della mammella (in particolare soprattutto carcinomi duttali invasivi) aumentato di 3,5 volte rispetto alla popolazione generale [12]. Per tale motivo è indicato che le donne affette da NF1 eseguano controlli senologici/mammografici annuali a partire dai 40 anni. Il trattamento non differisce da quello utilizzato nei casi sporadici.
Carcinoidi (o tumori neuroendocrini): si tratta di neoplasie neuroendocrine che originano dalle cellule della mucosa o sottomucosa del tratto gastrointestinale o dei polmoni, ma possono coinvolgere anche il mediastino, il timo, il pancreas, il fegato, i reni, i testicoli, le ovaie o la prostata. Colpiscono circa l’1% dei soggetti con NF1 e coinvolgono quasi esclusivamente la regione duodenale periampullare. Nella maggior parte dei casi si tratta di somatostatinomi. Generalmente si presentano con ittero, dolore addominale aspecifico, calo ponderale; meno comuni sono la melena, l’anemia da carenza di ferro, l’ostruzione intestinale e la pancreatite; la sindrome da somatostatinoma (diabete, diarrea e calcoli biliari) è estremamente rara e il quadro clinico può rispondere alla terapia con analoghi. Il trattamento consiste nella resezione chirurgica, ove possibile.
Tumori dei glomi subungueali
Pur trattandosi di una condizione benigna, costituiscono una problematica di grande impatto nella vita quotidiana a causa della loro sintomatologia importante, che spesso viene sottovalutata, e la cui diagnosi è ancora troppo tardiva. I tumori dei glomi subungueali sono neoformazioni rare nella popolazione generale - rappresentando meno del 2% delle neoplasie della mano – certamente più frequenti nei soggetti affetti da NF1, sebbene ad oggi l’incidenza non sia ancora definita con precisione.
I tumori glomici sono costituiti da un’iperplasia benigna delle cellule mioepitelioidi dei corpi glomici, ossia unità funzionali arterovenose precapillari deputate alla termoregolazione vascolare, localizzati a livello del tessuto sottocutaneo soprattutto a livello delle falangi distali delle dita, in particolare al di sotto del letto ungueale.
Si manifestano come neoformazioni a lenta crescita, di dimensioni variabili da alcuni millimetri ad alcuni centimetri (le tumefazioni più rilevanti possono manifestarsi come deformità della placca ungueale), talvolta di colorito rosso-violaceo; spesso sono multifocali, a differenza dei casi sporadici [13].
Il sintomo cardine è il dolore acuto localizzato al polpastrello o alla base dell’unghia corrispondente, spontaneo o correlato a stimoli meccanici anche in seguito a traumi di lieve entità; tipicamente il dolore aumenta con le basse temperature e diminuisce con il calore o esercitando una compressione alla base del dito.
Queste lesioni sono il paradigma di come la conoscenza delle complicanze e dei loro sintomi sia fondamentale per una corretta presa in carico dei pazienti. Molto spesso chi ha sviluppato tumori dei glomi ha alle spalle anni di complessi iter diagnostici strumentali e di inutili terapie mediche (antidolorifici, antidepressivi, antinfiammatori…). Tuttavia i sintomi sono talmente specifici e tipici, che la diagnosi clinica è immediata e quasi di certezza, anche se deve essere accompagnata dalla diagnostica per immagini (radiografia, ecografia, e risonanza magnetica nucleare, in alcuni casi) per la precisa localizzazione delle lesioni. La terapia è chirurgica e usualmente risolutiva sui sintomi [14].
Ringraziamenti
Gli autori desiderano ringraziare tutti i colleghi dell’IRCCS Fondazione Ospedale Maggiore Policlinico di Milano che da anni collaborano nella diagnosi e follow up dei pazienti affetti da NF1; i colleghi dell’Istituto Neurologico C. Besta, UO Neurooncologia Molecolare, dell’Istituto dei Tumori di Milano, UO Chirurgia Sarcomi e UO Oncologia Medica Tumori Mesenchimali dell’Adulto e Tumori Rari, e i colleghi dell’Università degli Studi di Milano, Dipartimento di Biotecnologie Mediche e Medicina Traslazionale. Infine un ringraziamento particolare all’ANF Associazione Neurofibromatosi Onlus.
Bibliografia
- Kayes LM, Burke W, Riccardi VM et al. Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet. 1994; 54:424-36.
- Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140-51.
- Pinna V, Lanari V, Daniele P et al. p.Arg-1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015; 23:1068–71.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-16.
- Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001; 68:1110-8.
- Gutmann DH, Ferner RE, Listernick RH et al. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017; 23;3:17004.
- Listernick R, Louis DN, Packer RJ et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41:143-9.
- Shamji MF, Benoit BG. Syndromic and sporadic pediatric optic pathway gliomas: review of clinical and histopathological differences and treatment implications. Neurosurg Focus. 2007;23(5):E3.
- Ueoka DI, Nogueira J, Campos JC et al. Brainstem gliomas-retrospective analysis of 86 patients. J Neurol Sci. 2009;281(1-2):20-3.
- Evans DG, Baser ME, McGaughran J et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-4.
- Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002. 15(6):679-84.
- Sharif S, Moran A, Huson SM et al. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007; 44(8):481-4.
- Brems H, Park C, Maertens O et al. Glomus tumors in neurofibromatosis type 1: genetic, functional, and clinical evidence of a novel association. Cancer Res. 2009; 15;69(18):7393-401.
- Morey VM, Garg B, Kotwal PP. Glomus tumours of the hand: Review of literature. J Clin Orthop Trauma. 2016; 7(4):286-291.