




la Newsletter

Giulia Luraschi1, Angelo Selicorni2
1Ospedale “F. Del Ponte”, Università dell’Insubria, Varese
2UOC di Pediatria, Centro Fondazione Mariani per il Bambino Fragile, ASST Lariana Como
La sindrome CHARGE | Coloboma, difetti cardiaci, atresia delle...
Coloboma, difetti cardiaci, atresia delle coane, ritardo di...
Gioele ha 8 mesi. Il suo peso alla nascita si attestava fra il 25° e il 50° percentile e ha mantenuto una crescita costante. Riscontro all’ecografia prenatale di arco aortico destro. Alla nascita, note dismorfiche al viso: deviazione a sx della rima orale durante il pianto, incompleta chiusura dell’occhio sx, padiglioni auricolari squadrati e a coppa.
Diagnosticata una ipoacusia trasmissiva destra e neurosensoriale sinistra per cui si decide di eseguire una valutazione oculistica con riscontro di coloboma corioretinico bilaterale e lagoftalmo monolaterale. Si effettua dunque una RMN encefalo che documenta una ipoplasia dei canali semicircolari. Nel sospetto di sindrome CHARGE, si procede ad analisi genetica NGS che rileva una variante in eterozigosi dell’introne 25 del gene CHD7 (variante patogenetica). In Gioele sono assenti le alterazioni ORL tipiche del quadro CHARGE e i disturbi del sonno così come le alterazioni dell’alimentazione. Dal punto di vista del neurosviluppo riscontro di ipotonia generalizzata, prevalentemente assiale, motricità spontanea ridotta, deficit di fissazione dello sguardo e dell’inseguimento visivo.
La sindrome CHARGE
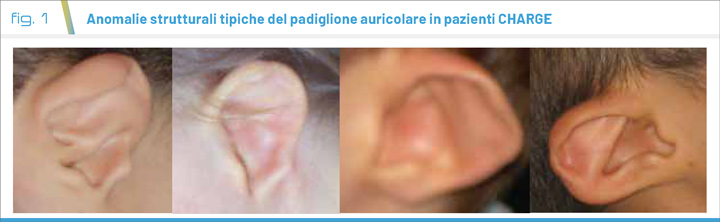
La sindrome CHARGE è una sindrome malformativa derivante nella maggior parte dei casi da mutazioni di una delle due copie del gene CHD7 (1). Il termine CHARGE è un acronimo delle caratteristiche principali: coloboma oculare (Coloboma), difetti cardiaci (Heart defect), atresia delle coane (Atresia choanae), ritardo della crescita e/o dello sviluppo (Retarded of growth and development), malformazioni genitourinarie (Genital hypoplasia), anomalie dell'orecchio (Ear anomalies) funzionali (ipoacusia neurosensoriale) e strutturali a carico dei padiglioni auricolari (2) (Fig. 1). L’incidenza è stimata tra 1/8500 e 1/15000 nati (3). La mortalità infantile è significativa quando presenti malformazioni cardiache, del sistema nervoso centrale, delle coane o atresia esofagea (4). In assenza di tali malformazioni la sopravvivenza non è significativamente compromessa; sono noti pazienti che hanno raggiunto l’età adulta (5).
Elementi di sospetto
Le malformazioni descritte nell’acronimo rappresentano i primi elementi di sospetto. I pazienti possono però giungere all’osservazione clinica anche in relazione a problemi di crescita e/o di sviluppo psicomotorio. Di seguito, i tratti somatici caratteristici: viso squadrato con diametro bifrontale stretto, naso largo, bocca piccola, labbro superiore invertito a forma di “V”, padiglioni auricolari squadrati o a coppa, anteversi con lobulo assente e, spesso, con elice sottile. Tipica la piega palmare a forma di bastone da hockey. Quasi costanti le anomalie dei nervi cranici che rendono ragione della paralisi del nervo facciale (5).
Criteri diagnostici
La diagnosi di sindrome CHARGE è clinica, basata sulla presenza di criteri maggiori e minori (6). I criteri maggiori sono rappresentati da Coloboma, atresia delle Coane, Canali semicircolari ipoplasici (identificati come le 3C); i minori includono anomalie del rombencefalo, disfunzioni ipotalamo-ipofisarie, malformazioni dell’orecchio esterno/medio e degli organi mediastinici, ritardo psicomotorio/cognitivo (2). Il riscontro dei tre criteri maggiori è necessario e sufficiente per porre diagnosi. Si parla di sindrome CHARGE anche in presenza di 2 elementi maggiori e 3 criteri minori. Vi sono inoltre un fenotipo CHARGE parziale (due criteri maggiori e nessuno minore) ed uno atipico (un criterio maggiore e almeno tre minori) (6).
Auxologia
I pazienti affetti hanno di solito un peso e una lunghezza adeguati alla nascita, ma la maggior parte va incontro a scarsa crescita (2), in conseguenza alle problematiche nutrizionali/deglutitorie e ai difetti cardiaci. Meno frequente (10%) il riscontro di deficit di GH (5).
Malformazioni e complicanze mediche associate
La malformazioni congenite più frequenti sono le cardiopatie (discreta prevalenza dei difetti tronco-conali), le anomalie oculari (coloboma), i difetti coanali (atresia/stenosi), la labio-palatoschisi (20-30%), l’atresia esofagea con o senza fistola tracheo-esofagea, le anomalie genitali (micropene e criptorchidismo nel maschio; anomalie uterine e delle labbra nelle femmine), le anomalie renali (disgenesia renale, rene a ferro di cavallo e doppio distretto) e le anomalie del sistema nervoso centrale (5). I disturbi dell’alimentazione (deficit della suzione, difficoltà di deglutizione, aspirazione cronica, reflusso gastro-esofageo) sono una tipica complicanza, conseguenza delle anomalie dei nervi cranici; spesso vi è necessità di alimentazione enterale. Frequente la presenza di deficit uditivo (neurosensoriale e/o trasmissivo) (2). Il riscontro di canali semicircolari assenti o ipoplasici comporta la presenza di disturbi dell’equilibrio da tenere in considerazione nella gestione quotidiana mentre l’ipoplasia dei bulbi olfattivi è alla base dell’ipo-anosmia (5). La gravità e l’estensione del coloboma oculare condizionano l’acuità visiva. Comune la fotofobia. In caso di paralisi del nervo facciale sono possibili le lesioni corneali (2). Raro il deficit di GH (10%), mentre frequente è l'ipogonadismo ipogonadotropo condizionante ipoplasia genitale e ritardo puberale (5). In una minoranza di pazienti viene riscontrato ipotiroidismo (10-15%) e nel 30% circa dei casi vi è insorgenza di epilessia. Riportate anomalie dentali (oligodontia, ritardo di eruzione dentaria). Anche il sonno può essere alterato (2).
Sviluppo psico-intellettivo
Presente un ritardo dello sviluppo psicomotorio di entità estremamente variabile e condizionata dalla presenza/gravità delle anomalie vestibolari e visive. Possibile compromissione dello sviluppo cognitivo; alcuni pazienti raggiungono dei valori di QI vicini alla normalità, altri presentano alterazioni intellettive significative. L’acquisizione del linguaggio è variamente ritardata, condizionata dalla gravità dei deficit sensoriali (5).
Difetto genetico
Il gene associato alla sindrome CHARGE è CHD7, localizzato sul braccio lungo del cromosoma 8 (8q12). Tale gene codifica per una proteina della famiglia delle Elicasi leganti il DNA, deputata alla regolazione dell’espressione genica. Gli effetti delle alterazioni di CHD7 dipendono dal tessuto in cui è espresso e dallo stadio della organogenesi (7). Il 90-95% dei pazienti che soddisfa i criteri diagnostici presenta una mutazione o una delezione eterozigote a carico di CHD7. La condizione presenta una trasmissione autosomica dominante e la maggioranza delle mutazioni sono eventi de novo. Descritte soprattutto mutazioni di tipo nonsense o frameshift, più rare le mutazioni missense, spesso con conseguenze cliniche più lievi (2).
Divulgazione di informazioni e sostegno
MONDO CHARGE (https://mondocharge.it/) è un’associazione fondata da persone con la sindrome CHARGE e dai loro familiari e ha come obiettivo principale quello di creare una rete di comunicazione e scambio d’informazioni fra i pazienti, le loro famiglie e gli operatori sociosanitari.
Bibliografia
- van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet C Semin Med Genet. 2017;175(4):397–406.
- Hsu P, Ma A, Wilson M, et al. CHARGE syndrome: A review: A review of CHARGE syndrome. J Paediatr Child Health. 2014;50(7):504–11.
- van Ravenswaaij-Arts CM, Hefner M, Blake K, Martin DM. CHD7 Disorder. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., curatori. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [citato 9 marzo 2023]. Disponibile su: http://www.ncbi.nlm.nih.gov/books/NBK1117/
- Issekutz KA, Graham JM, Prasad C, et al. An epidemiological analysis of CHARGE syndrome: Preliminary results from a Canadian study. Am J Med Genet A. 2005;133A(3):309–17.
- Tajè S, Cianci P, Selicorni A. La sindrome CHARGE. Medico E Bambino. 2017;9:585–6.
- Verloes A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am J Med Genet A. 2005;133A(3):306–8.
- Bergman JEH, Janssen N, Hoefsloot LH, et al. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48(5):334–42.