




la Newsletter

Alberto Budillon1, Martina Caiazza2, Giuseppe Limongelli2
1Dipartimento di Medicina di Precisione, Università della Campania “Luigi Vanvitelli”, Napoli. 2Unità di Malattie Rare e Genetiche Cardiovascolari, A.O.R.N. Dei Colli, Ospedale Monaldi, Napoli
La sindrome di Aymé-Gripp | Ritardo psicomotorio, cataratta,...
Ritardo psicomotorio, cataratta, sordità e facies caratteristica...
Presentiamo il caso di una bambina nata da parto naturale a termine con un peso di 2810 g (7° pc) ed immediatamente sottoposta ad una pericardiocentesi per il riscontro di un versamento pericardico. A quattro giorni di vita, viene trasferita nel reparto di terapia intensiva neonatale al fine di indagare più in profondità alcune caratteristiche fenotipiche e migliorare il suo regime alimentare.
Il versamento pericardico viene trattato con ibuprofene e furosemide per tre mesi. Le vengono quindi richiesti un cariotipo ed un array-CGH, entrambi risultati nella norma.
Durante l’infanzia, valutata dal punto di vista neuro-psichiatrico, le è stata suggerita psicomotricità e logopedia per il riscontro di ritardo del neurosviluppo. La visita oculistica e quella audiologica di screening sono risultate entrambe nella norma. A causa di una lieve scoliosi è stata sottoposta ad un Rx della colonna che ha mostrato la fusione degli archi posteriori delle vertebre C6-D7.
Dall’età di 6 anni, la paziente viene indirizzata al nostro dipartimento di Malattie Rare Cardiovascolari dell’Università della Campania “Luigi Vanvitelli” per una valutazione genetica e cardiologica.
La storia familiare risulta silente per condizioni genetiche. L’esame clinico rileva una facies piatta, naso piccolo, filtro lungo, bocca piccola, orecchie ruotate posteriormente e unghie distrofiche. L’esame cardiologico risulta invece nella norma. Esegue anche una RMN dell’encefalo che evidenzia una malformazione di Chiari di tipo I.
La sindrome di Aymé-Gripp
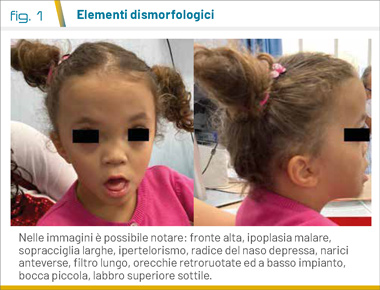 La sindrome di Aymé-Gripp (OMIM 601088) è una sindrome genetica ultrarara derivante nella maggior parte dei casi da mutazioni de novo missenso di una delle due copie del gene MAF (1). A partire dal 1983 Fine e Lubinsky raccolsero una casistica di pazienti con fenotipo simile: brachicefalia, cataratta bilaterale ad esordio precoce, sordità neurosensoriale e volto peculiare in combinazione con ritardo del neurosviluppo erano presenti ciascuno in più del 90% dei pazienti (2). La facies è spesso descritta come Down-like ed include volto con ipoplasia della regione mediana, ipertelorismo, radice del naso larga, naso corto, narici anteverse, filtro lungo, bocca piccola, labbro superiore sottile, orecchie a basso impianto e retroruotate, anomalie della dentizione (Fig. 1). Sono anche riportate bassa statura, ipoplasia delle ghiandole mammarie, anomalie ectodermiche come capelli radi e unghie distrofiche, sinostosi radio-ulnare, crisi convulsive, malformazione di Chiari tipo I e versamento pericardico (3). Ad oggi sono stati descritti meno di 30 pazienti (4).
La sindrome di Aymé-Gripp (OMIM 601088) è una sindrome genetica ultrarara derivante nella maggior parte dei casi da mutazioni de novo missenso di una delle due copie del gene MAF (1). A partire dal 1983 Fine e Lubinsky raccolsero una casistica di pazienti con fenotipo simile: brachicefalia, cataratta bilaterale ad esordio precoce, sordità neurosensoriale e volto peculiare in combinazione con ritardo del neurosviluppo erano presenti ciascuno in più del 90% dei pazienti (2). La facies è spesso descritta come Down-like ed include volto con ipoplasia della regione mediana, ipertelorismo, radice del naso larga, naso corto, narici anteverse, filtro lungo, bocca piccola, labbro superiore sottile, orecchie a basso impianto e retroruotate, anomalie della dentizione (Fig. 1). Sono anche riportate bassa statura, ipoplasia delle ghiandole mammarie, anomalie ectodermiche come capelli radi e unghie distrofiche, sinostosi radio-ulnare, crisi convulsive, malformazione di Chiari tipo I e versamento pericardico (3). Ad oggi sono stati descritti meno di 30 pazienti (4).
Difetto genetico
Il gene MAF (OMIM 177075) è mappato sul cromosoma 16q23 e codifica per un fattore di trascrizione coinvolto nel differenziamento cellulare dei linfociti T helper-2. Questa proteina gioca un importante ruolo nella regolazione di diversi processi cellulari come nello sviluppo delle cellule embrionali del cristallino (5). Alcune sostituzioni amminoacidiche in MAF causano la sindrome di Aymé-Gripp, altre sono responsabili della sua forma allelica caratterizzata da cataratta isolata (OMIM 610202). Queste due possibili condizioni si realizzano alternativamente quando le varianti patogenetiche cadono in uno dei due cluster descritti da Niceta et al. (6).
Elementi di sospetto clinico
Le caratteristiche descritte dalla tetrade rappresentano i primi elementi di sospetto, ma i pazienti possono giungere all’osservazione clinica anche in relazione a problemi di crescita. La nostra paziente presenta numerose delle caratteristiche note della sindrome di Aymé-Gripp, e tra tutte il quadro dismorfologico predomina. Tuttavia, mancano due elementi della tetrade presenti nella maggior parte dei casi riportati: la cataratta bilaterale ad esordio precoce e la sordità neurosensoriale, mai assenti insieme negli altri casi.
Indagine genetica
Dal sequenziamento dell’esoma clinico effettuato in trio viene evidenziata una variante de novo missenso in MAF (NM_005360.5: c.161C>G; p.Ser54Trp) classificata come likely pathogenic secondo i criteri ACMG (American College of Medical Genetics) e già precedentemente descritta in letteratura in pazienti presentanti un fenotipo classico. Viene quindi interpretata come causativa della condizione della paziente.
Considerazioni conclusive
In assenza di cataratta e sordità neurosensoriale, la nostra risulta essere la prima paziente descritta con un fenotipo meno severo. Con l’avvento di metodi di sequenziamento sempre più performanti, ci si aspetta di scoprire mutazioni causative in pazienti con manifestazioni sempre più sfumate rispetto al passato, soprattutto nell’ambito delle malattie rare. Risulterà perciò sempre più importante sospettare una condizione genetica anche in assenza di alcune varianti fenotipiche considerate segni “maniglia”.
Bibliografia
- Wang Q, Qin T, Tan H, et al. Broadening the genotypic and phenotypic spectrum of MAF in three Chinese Han congenital cataracts families. Am J Med Genet A. 2022;188(10):2888-2898.
- Aymé S, Philip N. Fine-Lubinsky syndrome: a fourth patient with brachycephaly, deaf-ness, cataract, microstomia and mental retardation. Clin Dysmorphol.1996; 5(1):55-60.
- König AL, Sabir H, Strizek B, et al. Isolated cytokine-enriched pericar-dial effusion: A likely key feature for Aymé-Gripp syndrome. Am J Med Genet A. 2022; 188(2):624-627.
- Amudhavalli SM, Gadea R, Gripp K. Aymé-Gripp Syndrome. 2020 Feb 6. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA).
- Amudhavalli SM, Hanson R, Angle B, et al. Further delineation of Aymé-Gripp syndrome and use of automated facial analysis tool. Am J Med Genet A. 2018; 176(7):1648-1656.
- Niceta M, Stellacci E, Gripp KW, et al. Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am J Hum Genet. 2015;96(5):816-25.