




la Newsletter
La sindrome di Lynch raccontata dalla Associazione AIFEG | Il...
Il riconoscimento della sindrome di Lynch nell’ambito...
Tra le novità introdotte dai Livelli Essenziali di Assistenza (LEA) nel 2017 figura anche l’inserimento tra le Malattie Rare di una delle forme più note di predisposizione al cancro nell’adulto: la sindrome di Lynch (SL), responsabile della maggior parte dei tumori colorettali ereditari (1).
Il cancro colorettale (CCR) è uno dei tumori più frequenti nella popolazione adulta occidentale: in Italia rappresenta nei maschi il 15% e nelle femmine il 13% di tutti i tumori (2). La maggior parte dei casi si presenta in forma sporadica, anche se il 15-20% dei pazienti riferisce una familiarità di primo grado. Oltre che del 3% dei CCR, la sindrome è responsabile anche del 2% dei tumori all’endometrio (2,3). Complessivamente, a seconda delle diverse popolazioni, la sua incidenza viene valutata in 1:660-2000 (4).
Dal punto di vista molecolare, la SL è associata a mutazioni a carico di uno dei geni codificanti le proteine coinvolte nel sistema di riparazione del DNA (Mismatch Repair, MMR), trasmesse con modalità autosomico dominante (AD) (Fig. 1).
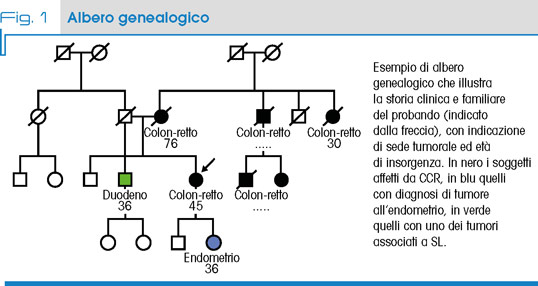
Il riconoscimento della SL nell’ambito dell’elenco nazionale delle Malattie Rare è stato di particolare importanza perché permette l’accesso alle facilitazioni assistenziali che ne derivano non solo ai pazienti ma anche ai loro familiari portatori di mutazione. Questo, grazie al progressivo espandersi dell’uso diagnostico dei pannelli genici, sta portando ad un aumento dell’identificazione di nuovi casi, ridisegnando i dati di prevalenza della sindrome.
Diagnosi
I criteri di sospetto diagnostico sono schematizzati nelle tabelle 1 e 2. I criteri di Amsterdam (Tab. 1), si rifanno essenzialmente alla storia clinica e familiare dei pazienti.
.jpg)
Successivamente, questi criteri sono stati integrati dai criteri di Bethesda (Tab. 2) che considerano anche l’instabilità dei microsatelliti e del difetto di espressione di almeno una delle proteine del sistema del MMR. In questa classificazione assumono un peso maggiore l’età di insorgenza del cancro, l’istologia e le caratteristiche genetiche dei CCR, nonché la presenza di altri tumori oltre a quelli strettamente associati alla sindrome (5).

La diagnosi è accertata quando i test genetici identificano una mutazione patogenetica in uno dei geni MMR; tuttavia esistono storie familiari caratterizzate da ricorrenza di tumori gastroenterici e dell’endometrio che simulano la SL, ma in cui la mutazione patogenetica non è identificabile. Potrebbe trattarsi di famiglie in cui sono presenti mutazioni di geni diversi o alterazioni di tipo epigenetico. Le indicazioni recenti dell’Associazione Italiana di Oncologia Medica (AIOM) raccomandano per tutti i pazienti, che rientrano nei criteri, una consulenza genetica oncologica per la corretta identificazione delle sindromi ereditarie con i test adeguati (6). Questo aspetto è particolarmente rilevante per attivare i programmi di prevenzione rivolti ai portatori asintomatici.
Genetica
La SL è trasmessa con un’ereditarietà AD, ed è dovuta a mutazioni costituzionali in eterozigosi di uno dei geni del sistema MMR: MSH2, MLH1, MSH6, PMS2 e EPCAM (quest’ultimo, pur non appartenendo al MMR, è un gene la cui delezione causa l’inattivazione del gene adiacente, MSH2). Le mutazioni patogenetiche di questi geni predispongono allo sviluppo di tumori prevalentemente colorettali e dell’endometrio, ma, in misura minore, anche di tumori in altre sedi: ovaio, stomaco, rene e ureteri, piccolo intestino, cervello, cute, pancreas e vie biliari.
Il processo di cancerogenesi inizia con un classico meccanismo di “second hit”: una seconda mutazione somatica, nell’allele non mutato costituzionalmente, inattiva completamente il gene con conseguente perdita della proteina corrispondente. Ciò, a sua volta, fa perdere alla cellula la capacità di contrastare l’insorgenza di errori lungo il DNA e quindi anche di mutazioni somatiche in altri proto-oncogeni e/o geni oncosoppressori.
I 4 geni responsabili della SL (MSH2, MSH6, MLH1 e PMS2) hanno diversa penetranza ed espressività. Il rischio maggiore per CCR è legato a mutazioni in MSH2 e MLH1. Il rischio cumulativo entro i 70 anni per questo tipo di tumore arriva al 46% e 35%, rispettivamente per i portatori di mutazioni di MLH1 e MSH2. Il rischio è minore per mutazioni di MSH6, 20%, e PMS2, 10%.
Per le donne, si aggiunge il rischio per il tumore all’endometrio, la cui incidenza è alta soprattutto tra le portatrici di mutazioni di MSH2 (51%), MSH6 (49%) e MLH1 (34%). Il tumore è frequente anche nelle donne anziane portatrici di mutazioni di PMS2. Va segnalato, infine, l’alto rischio per il tumore dell’ovaio, con incidenza variabile tra l'11% e il 15%.
Da questi dati deriva quindi un rischio complessivo sino al 75% di sviluppare uno dei tumori dello spettro SL (3).
In rari casi, con ereditarietà autosomico recessiva (AR), è stata riscontrata la presenza di due varianti costituzionali: i due alleli dello stesso gene sono entrambi mutati (mutazione biallelica). La condizione è conosciuta come “Constitutional Mismatch Repair Deficiency” (CMMRD) e si presenta già in età infantile o giovanile, con un quadro clinico severo comprendente: tumori cerebrali, adenomi e adenocarcinomi colici e intestinali, tumori all’endometrio, linfomi, leucemie, e macchie cutanee “caffelatte” (caratteristica confondente con la neurofibromatosi tipo 1). Nella metà dei pazienti è stata riscontrata una situazione familiare di consanguineità. I geni più coinvolti sono PMS2 e MSH6 (7).
Test genetici
Test su DNA tumorale
La mancanza dell’espressione di una delle proteine del MMR nel tessuto tumorale è uno degli elementi di sospetta SL, anche in assenza di familiarità.
L’analisi immunoistochimica delle proteine coinvolte (MSH2, MSH6, MLH1 E PMS2) è quindi un test strategico per l’identificazione della sindrome (8), disponibile in tutti i reparti di Anatomia Patologica.
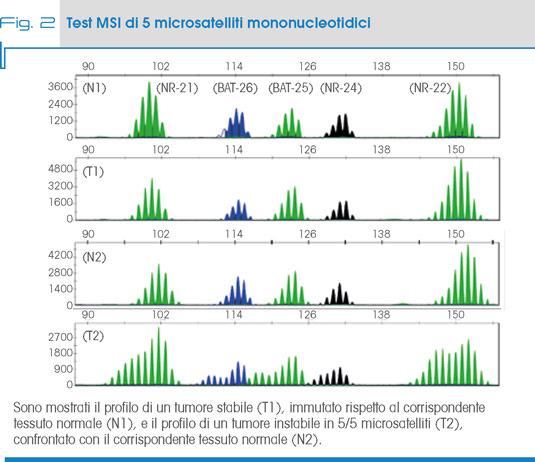
Invece, sull’instabilità nei microsattelliti (MSI) si basa il test di pre-screening elettivo che analizza un pannello di 5 microsatelliti mononucleotidici: BAT25, BAT26, NR21, NR22, NR24. L’instabilità è definita alta in presenza di 2-5 microsatelliti instabili (High, MSI-H), bassa in presenza di 1 microsatellite instabile (Low, MSI-L) o assente (Stable, MSS), (Fig. 2).
La presenza di MSI può coinvolgere anche tumori sporadici, in seguito all’inattivazione somatica di un gene del MMR. In particolare, MLH1 può presentare inattivazione epigenetica secondaria ad ipermetilazione del promotore genico (9). La metilazione di MLH1 va quindi sempre indagata nei tumori non esprimenti la proteina MLH1, insieme alla ricerca della mutazione somatica BRAF V600E correlata.
La presenza di ipermetilazione somatica e/o di mutazione BRAF esclude la SL, così come una mutazione biallelica somatica di uno dei quattro geni.
I test somatici della mutazione V600E di BRAF e il test di metilazione del promotore di MLH1 rappresentano un importante supporto per accertare la natura sporadica di una neoplasia con difetto dei geni MMR.
Test su DNA costituzionale (da sangue)
L’analisi molecolare della parte codificante del gene interessato va richiesta dopo aver effettuato l’accertamento dell’instabilità dei microsatteliti e/o la presenza di un’immunoistochimica difettiva. Il test va completato con lo studio dell’eventuale presenza di grandi delezioni o duplicazioni, coinvolgenti uno o più esoni, mediante Multiple Ligation- dependent Probe Amplification (MLPA).
Oggi molti laboratori di genetica si stanno inoltre rapidamente convertendo alla tecnologia della “Next Generation Sequencing (NGS)” che permette l’analisi simultanea di molti geni, anche mediante l’uso di kit commerciali o custom per l’amplificazione e sequenza di pannelli genici selezionati.
La risposta del laboratorio di genetica dovrebbe riportare tutte le varianti genetiche non sicuramente innocue trovate nel DNA del paziente, specificando la tecnica utilizzata e la sensibilità della stessa. Allo stesso tempo dovrebbe riportare l’interpretazione delle varianti alla luce delle attuali conoscenze.
Per la classificazione delle varianti, l’International Society for Gastrointestinal Hereditary Tumours (InSi- GHT) raccomanda uno schema a cinque classi, riportata in tabella 3. I dati clinici, molecolari, di predizione in silico e di segregazione della variante concorrono a definirne la classe di appartenenza. Il database di riferimento per le varianti dei geni del MMR è l’InSiGHT variant database (insight-database.org), che riporta le varianti identificate e (in parte) classificate secondo questi criteri. Una volta identificata una mutazione patogenetica la ricerca va allargata ai familiari del paziente.

Sorveglianza e trattamento
La gestione clinica dei pazienti con SL e degli individui sani a rischio dovrebbe considerare tutti i tumori dello spettro, come indicato dalle Linee Guida dell’European Hereditary Tumour Group (10), dell’American College of Gastroenterology (11) e del National Comprensive Cancer Network (12).
La sorveglianza per CCR tramite colonscopia dovrebbe essere effettuata ogni 1-2 anni, a partire dai 20-25 anni. La colectomia completa con ileo retto-anastomosi (IRA) e la colectomia sub-totale sono i trattamenti d’elezione per pazienti con CCR, ma anche per quelli con neoplasie coliche benigne non facilmente controllabili con endoscopia. Per le donne con mutazione accertata è indispensabile anche la sorveglianza ginecologica, con ecografia trans vaginale annuale, a partire dai 30-35 anni.
L’isterectomia e la ovariectomia profilattica possono essere valutate e offerte alle donne a conclusione della vita riproduttiva e generalmente intorno ai 40 anni. Riguardo al rischio degli altri tumori, non c’è ancora ampio consenso e la sorveglianza andrebbe condotta all’interno di protocolli di ricerca. Oltre allo screening annuale per H. pylori su tutti i portatori di mutazione, l’endoscopia del tratto gastrointestinale superiore (gastroscopia e duodenoscopia) è suggerita ogni 2-3 anni, per lo più nei paesi con alta incidenza di cancro gastrico e/o nelle famiglie con tumori in questi distretti. La sorveglianza mediante citologia urinaria è invece indicata a partire dai 30-35 anni soprattutto nei portatori di mutazione nel gene MSH2.
Attualmente sono in corso di valutazione studi di chemioprevenzione con aspirina.
Va infine precisato che, al di là della sorveglianza intensiva, la gestione clinica dei pazienti con SL deve essere personalizzata in base al dato genetico e alle caratteristiche cliniche, personali e familiari. Non da ultimo, chi si fa carico dei pazienti deve essere consapevole degli aspetti psicologici e relazionali, prevedendo un eventuale supporto specifico.
Sindrome di Lynch: la parola agli esperti
I marcatori: dall’istopatologia alla patologia molecolare
Matteo Fassan
L’esame istopatologico può rilevare alcune caratteristiche peculiari del tumore che suggeriscono la presenza di un difetto del MMR: (i) uno scarso grado di differenziazione delle cellule neoplastiche (ove il grado di differenziazione definisce quanto la neoplasia assomiglia al tessuto sano di partenza); (ii) la presenza di aree tumorali con morfologia di infrequente evenienza quali l’istotipo mucinoso o a cellule con castone (signet ring cells); (iii) un istotipo midollare definito con i criteri della classificazione mondiale della sanità (WHO 2010) come neoplasia scarsamente differenziata con pattern di crescita solido ed infiltrato infiammatorio prominente; (iv) presenza di un infiltrato infiammatorio linfocitario intratumorale di alto grado; (v) presenza di una reazione infiammatoria peritumorale simile a quella osservata in pazienti con morbo di Crohn; (vi) presenza di eterogeneità fenotipica del tumore. Tuttavia, una importante frazione di tumori con difetti del MMR non presenta tali caratteristiche. Questo costituisce la base biologica allo screening universale applicato a tutte le neoplasie coliche resecate (8).
Da un punto di vista patogenetico, gli adenocarcinomi insorti in SL possono originare da lesioni adenomatose e sono quindi caratterizzati da alterazioni solitamente descritte nella sequenza adenoma-carcinoma, quali difetti nei geni APC, CTNNB1 o KRAS, mentre sono estremamente rare le mutazioni a carico del gene BRAF.
Algoritmo diagnostico e nuove strategie
Maria Grazia Tibiletti
Poiché i tumori dello spettro della SL presentano come caratteristiche biologiche comuni difetti del sistema del MMR, considerata l’eterogeneità genetica che caratterizza la sindrome, i test genetici sul tumore (somatici) rappresentano un’importante risorsa per identificare i pazienti da sottoporre a test genetici costituzionali. L’algoritmo riconosciuto a livello internazionale per identificare la SL prevede infatti le analisi somatiche che comprendono i test MSI e di espressione immunoistochimica delle quattro proteine del MMR e successivamente le analisi costituzionali mirate sui geni risultati difettivi. In presenza di difetto concomitante di MLH1 e PMS2 vengono utilizzati gli altri test somatici (mutazione di BRAF e metilazione del promotore del gene MLH1) per escludere dal test genetico costituzionale i tumori sporadici con difetto di MLH1. Recentemente è stato promosso un Network internazionale (13), per l’identificazione della SL che prevede l’uso del test immunoistochimico sui tumori colorettali ed endometriali come test universale per selezionare a tutte le età i pazienti con un sospetto di SL. Anche in Italia alcune regioni stanno promuovendo l’uso del test universale; è noto infatti dalla letteratura che circa il 50% dei pazienti Lynch non soddisfano i criteri di Amsterdam e che l’uso dei test universali rispetto ai criteri clinici incrementa di 2 volte l’identificazione dei pazienti Lynch (14).
Prospettive terapeutiche
Luca Roncucci
E’ ormai da tempo ben documentata la migliore sopravvivenza dei pazienti con CCR con deficit del sistema del MMR, rispetto ai pazienti con CCR senza tale deficit (15). Recentemente, tuttavia si è osservato che la chemioterapia adiuvante, dopo intervento chirurgico, può portare un ulteriore vantaggio in termini di sopravvivenza ai pazienti con SL. Ma non tutti i regimi terapeutici sono efficaci. Al contrario di fluorouracile (16), l’oxaliplatino sembra il chemioterapico più efficace. In particolare, si è visto che i pazienti con tumori con deficit del sistema del MMR in stadio III hanno una prognosi migliore quando trattati oltre che con fluoropirimidina, anche con oxaliplatino (17). L’impiego di strategie immunologiche per combattere le neoplasie ha da tempo impegnato diversi gruppi di ricerca in tutto il mondo. Un’azione delle terapie immunologiche sembra più efficace negli stadi premaligni delle neo- plasie, piuttosto che negli stadi più avanzati, con lo scopo fondamentale di prevenire lo sviluppo neoplastico. Gli studi in questo ambito sono ancora in gran parte preliminari; tuttavia, è stato di recente ipotizzato che la valutazione del profilo molecolare dei tumori, ed in particolare il deficit del MMR, possa aiutare nel giudicare il possibile beneficio clinico di terapie inibitorie su checkpoint immunitari della cellula (18). Nell’ambito di chemioprevenzione della cancerogenesi colorettale, le sostanze maggiormente studiate sono state antiossidanti, vitamine ed antiinfiammatori non steroidei. Un trial clinico (studio CAPP2) ha saggiato l’uso dell’ acido acetilsalicilico (ASA) per prevenire il CCR nei pazienti portatori di mutazioni nei geni del MMR. L’ASA alle dosi di 600 mg/die sembra portare ad un lieve effetto di riduzione del rischio di sviluppare il CCR, che risulta evidente però solo dopo 4 anni dall’inizio del trattamento (19). E’ in corso di avvio un ulteriore trial (CAPP3) per cercare di definire la dose ottimale di ASA a fini chemiopreventivi nella SL.
Chirurgia personalizzata
Antonio Chiappa
Le principali linee guida nazionali ed internazionali individuano la colectomia totale con eventuale conservazione del retto (se non sede di malattia) e con confezionamento di anastomosi ileo-anale, il trattamento chirurgico di elezione nei pazienti con diagnosi di CCR insorto in soggetti con SL (20). Il rischio di recidiva metacrona aumenta di 1,5% ogni anno rispetto alla popolazione generale sino ad un rischio cumulativo del 40-45%. La scelta di una chirurgia maggiormente conservativa, con la resezione segmentaria del solo tratto colico-rettale sede di insorgenza della neoplasia ed un regime di stretto follow up (colonscopia ed imaging annuale) non mostra, dai dati riportati, la stessa efficacia della chirurgia estesa-maggiore, a causa delle possibili variabili connesse all’esame endoscopico (esperienza dell’operatore, preparazione intestinale, ritardi nell’esecuzione dell’esame) e del rischio di sviluppo di carcinomi intervallari riportati nel 35% dei casi. Il paziente con diagnosi di SL andrà tuttavia adeguatamente edotto sull’impatto che un intervento maggiore come la colectomia totale (spesso eseguita in due o tre tempi chirurgici) potrà avere sulla sua qualità di vita, a fronte di un rischio di sviluppo di carcinoma che potrebbe rimanere solo teorico. Per quanto la resezione estesa rimanga il trattamento principe suggerito, è possibile individuare una “chirurgia di precisione” modellata sulle caratteristiche genotipiche e biologiche del singolo caso. La possibilità di individuare un vero e proprio pattern biologico, istologico e genotipico degli adenocarcinomi insorti in pazienti con SL può costituire infatti un criterio di scelta degli individui che maggiormente beneficerebbero di una resezione estesa e di casi a minore rischio che possano essere sottoposti a chirurgia conservativa e stretta sorveglianza. Infine, per quanto riguarda i tumori a sede extracolica, il trattamento chirurgico segue sempre i criteri adottati per la popolazione normale.
Il gold standard della sorveglianza endoscopica
Mara Fornasarig
La sorveglianza endoscopica nella SL è alla base della prevenzione delle neoplasie coliche nei portatori asintomatici di mutazioni e nei pazienti già operati per CCR. La lesione precancerosa colica è rappresentata dal polipo adenomatoso. La storia naturale del polipo adenomatoso che si sviluppa nell’ambito della sindrome ha la caratteristica di una rapida cancerizzazione. E’ stato stimato un tempo, fra la formazione del polipo e la sua trasformazione in cancro, di circa 2-3 anni, tempo nettamente inferiore rispetto a quello che si osserva nelle neoplasie coliche sporadiche e nella poliposi adenomatosa familiare. Inoltre, il rischio di lesioni metacrone è del 40-45%. La colonscopia rappresenta l’esame cardine per la diagnosi e l’asportazione dei polipi adenomatosi. L’esame deve essere effettuato da personale medico esperto e deve rispettare le caratteristiche di colonscopia di qualità previsto dalle comunità scientifiche. Per ridurre al minimo il numero di lesioni che possono sfuggire all’esame, viene suggerito di utilizzare strumenti endoscopici ad alta definizione o con luce NBI (narrow banding imaging), o la cromo endoscopia. E’ stato dimostrato che queste tecniche consentono di incrementare la percentuale di diagnosi di lesioni adenomatose (21). Dato che il tempo tra formazione e cancerizzazione di un polipo adenomatoso è breve, la cadenza dei controlli consigliata dalle linee guida è di 1-2 anni.
La gestione del paziente con un approccio multidisciplinare
Lupe Sanchez Mete
Nel percorso diagnostico-terapeutico della SL sono coinvolti vari specialisti: chi pone il sospetto diagnostico (gastroenterologo-endoscopista/genetista/oncologo), chi effettua i test diagnostici (anatomo-patologo e genetista) e chi è coinvolto nella sorveglianza oncologica e nell’eventuale trattamento sia dei portatori di mutazione asintomatici sia dei pazienti che hanno già sviluppato uno o più tumori (gastroenterologi endoscopisti, ginecologi, urologi, oncologi, chirurghi dell’apparato digerente, ecc)(10, 11). In questo contesto, il team multidisciplinare emerge come una necessità pratica per ottimizzare la comunicazione ed il coordinamento delle varie specialità coinvolte ed ha un impatto positivo sulla “detection rate” delle mutazioni, sull’aderenza al test genetico, e sulla “compliance” alle strategie di riduzione del rischio oncologico (22). Il ruolo del team multidisciplinare nella gestione dei pazienti affetti da malattia rara è inoltre riconosciuto dal Ministero della Salute quale Criterio di designazione e valutazione dei centri di “expertise” ed è inserito nel Percorso Diagnostico Terapeutico Assistenziale redatto dai Centri di Riferimento Regionali per ciascuna malattia rara. Negli ultimi 15-20 anni il “management” clinico dei pazienti oncologici si è modificato, passando da una gestione centrata sulla malattia ad una focalizzata e ritagliata sul singolo paziente (23). Secondo le attuali linee guida oncologiche nazionali ed internazionali (6, 12), il percorso di diagnosi e cura oncologica viene pianificato nell’ambito di un "disease management team" formalmente costituito, tumore-specifico, che prevede l’integrazione delle differenti professionalità (incluse cure palliative, scienze infermieristiche, psico-oncologia ecc.). Sono inoltre ormai chiare le evidenze dei benefici della gestione multidisciplinare che, in particolare, sembra avere un impatto favorevole sulla sopravvivenza.
Novità sull’esenzione e sulle Reti Malattie Rare
Renato Cannizzaro
Con il Decreto Ministeriale n. 279 del 18 maggio 2001 veniva istituita la Rete Nazionale delle Malattie Rare, riconoscendo l’esenzione dalla partecipazione al costo delle relative prestazioni sanitarie ai pazienti affetti da una o più malattie rare allo scopo di garantire a tutti i pazienti l’accesso ai diritti di: diagnosi, cura, esenzione. Con il Decreto Ministeriale del 15 aprile 2008 (G.U. Serie Generale n. 227 del 27 settembre 2008) venivano individuati i Centri interregionali per le malattie rare a bassissima prevalenza per razionalizzare la funzione dei presidi di riferimento e garantire l’erogazione delle cure il più vicino al domicilio del paziente. Successivamente a questi decreti, le legislazioni regionali hanno deliberato sulle Reti delle Malattie Rare. L’inserimento da parte del Ministero della Salute nei Livelli Essenziali di Assistenza 2017 (1) della SL ha permesso finalmente di usufruire dell’esenzione RBG021, che comprende tutte le prestazioni ambulatoriali correlate con la sindrome. La struttura di coordinamento regionale di rete assicura la presa in carico del paziente ed il completamento dell'iter diagnostico, terapeutico e di certificazione.
AIFEG: la ricerca e la cura in Italia
Guglielmina Nadia Ranzani, Presidente AIFEG
AIFEG (Associazione Italiana per lo studio della Familiarità ed Ereditarietà dei tumori Gastrointestinali), associazione italiana dedicata all’ottimizzazione di diagnosi e trattamento dei tumori ereditari del digerente, è stata ufficialmente fondata nel 2002 a Verona. Si tratta di un’associazione multidisciplinare che si pone come obiettivi specifici: l’identificazione dei fattori genetici che predispongono allo sviluppo dei tumori gastrointestinali; la prevenzione e il trattamento di tali tumori; la messa in atto di progetti collaborativi di ricerca di base, pre-clinica e clinica; lo scambio di conoscenze ed esperienze al fine di colmare il divario fra conoscenze di base e pratica clinica corrente; la più ampia diffusione delle conoscenze acquisite. In un contesto come quello italiano, non sempre pronto ad approcci multidisciplinari, AIFEG è diventata nel tempo un punto di riferimento per genetisti e clinici appartenenti a varie discipline, contribuendo a migliorare la gestione di pazienti e famiglie a rischio. Più recentemente, AIFEG sta supportando la nascita di un’associazione di pazienti con SL ed ha contribuito, interfacciandosi con il decisore politico in materia di sanità pubblica, ad ottenere esenzioni per test genetico e sorveglianza clinica per questa sindrome. AIFEG collabora, attraverso numerosi dei suoi soci, con società scientifiche italiane, con l’European Hereditary Tumour Group e con l’InSiGHT. La collaborazione con associazioni internazionali e con consorzi che raccolgono dati genetici e clinici si sta rivelando fondamentale in vista delle sfide che derivano dalle nuove tecniche di sequenziamento del DNA. Infatti, la possibilità di indagare un numero molto elevato di geni in pazienti selezionati per sospetta predisposizione ereditaria a tumori del colon- retto, dello stomaco, e dell’endometrio, sta portando alla luce un numero crescente di varianti in geni del tutto inattesi e di varianti a significato patogenetico incerto. Solo condividendo i risultati su larga scala sarà possibile migliorare la valutazione del rischio e selezionare l’informazione “actionable”, cioè da trasferire a pazienti e clinici in quanto utilizzabile per prevenire o modificare il decorso della malattia.
Bibliografia
- Gazzetta Ufficiale. www.gazzettaufficiale.it consultato 5 marzo 2018
- AIOM, AIRTUM eds. I numeri del cancro in Italia 2017. 2017th ed. ROMA: Il pensiero scientifico editore; 2017.
- Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 2017;66(3):464–472.
- de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer 2005;4(3):233–237.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J Natl Cancer Inst 2004;96(4):261–268.
- AIOM Linee Guide Tumore al colon. http://www.aiom.it/professionisti/documenti-scientifici/linee-guida/1,413,1 consultato 5 marzo 2018
- Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J Med Genet 2014;51(6):355–365.
- Remo A, Fassan M, Lanza G. Immunohistochemical evaluation of mismatch repair proteins in colorectal carcinoma: the AIFEG/GIPAD proposal. Pathologica 2016;108(3):104–109.
- Hitchins MP. The role of epigenetics in Lynch syndrome. Fam Cancer 2013;12(2):189–205.
- Vasen HFA, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013; 62(6):812–823.
- Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110(2):223–63.
- Benson AB, Venook AP, Al-Hawary MM, et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J Natl Compr Canc Netw 2018;16(4):359–369.
- Mange S, Bellcross C, Cragun D, et al. Creation of a network to promote universal screening for Lynch syndrome: the LynchSyndrome Screening Network. J Genet Couns 2015;24(3):421–427.
- Cohen SA, Laurino M, Bowen DJ, et al. Initiation of universal tumor screening for Lynch syndrome in colorectal cancer patients as a model for the implementation of genetic information into clinical oncology practice. Cancer 2016;122(3):393–401.
- Benatti P, Gafà R, Barana D, et al. Microsatellite instability and colorectal cancer prognosis. Clin Cancer Res Off J Am Assoc Cancer Res 2005;11(23):8332–8340.
- Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol Off J Am Soc Clin Oncol 2010;28(20):3219–3226.
- Tougeron D, Mouillet G, Trouilloud I, et al. Efficacy of Adjuvant Chemotherapy in Colon Cancer With Microsatellite Instability: A Large Multicenter AGEO Study. J Natl Cancer Inst 2016;108.
- Meng X, Huang Z, Teng F, Xing L, Yu J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat Rev 2015;41(10):868–876.
- Burn J, Gerdes A-M, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet Lond Engl 2011;378(9809):2081–2087.
- Herzig DO, Buie WD, Weiser MR, et al. Clinical Practice Guidelines for the Surgical Treatment of Patients With Lynch Syndrome. Dis Colon Rectum 2017;60(2):137–143.
- Rahmi G, Lecomte T, Malka D, et al. Impact of chromoscopy on adenoma detection in patients with Lynch syndrome: a prospective, multicenter, blinded, tandem colonoscopy study. Am J Gastroenterol 2015;110(2):288–298.
- Márquez-Rodas I, Lobo M, Flores-Sanchez C, et al. Five Years of Multidisciplinary Care in Hereditary Cancer: Our Experience in a Spanish University Hospital. Oncology 2017;92(2):68–74.
- European Partnership Action Against Cancer consensus group, Borras JM, Albreht T, et al. Policy statement on multidisciplinary cancer care. Eur J Cancer Oxf Engl 2014;50(3):475–480.