




la Newsletter

Marika Farioli1,2, Alessia Carrer2,3, Angelo Selicorni2
1Dipartimento donna e bambino, ASST-Sette Laghi, Università degli Studi dell’Insubria, Varese; 2UOC Pediatria. Presidio S. Fermo. ASST-Lariana, Como. Centro Fondazione Mariani per il Bambino Fragile; 3Università degli Studi di Milano – Scuola di Specializzazione in Genetica Medica
La sindrome di Niikawa-Kuroki | La sindrome di Niikawa-Kuroki,...
La sindrome di Niikawa-Kuroki, nota anche come sindrome Kabuki, è...
Gravidanza insorta tramite ovodonazione e complicata da polidramnios e riscontro di arteria ombelicale unica. Cariotipo fetale e analisi array-CGH su liquido amniotico nella norma. Nata a 35 settimane gestazionali da taglio cesareo urgente con peso neonatale di 2100 g, APGAR 9/10. Poche ore dopo la nascita comparsa di crisi ipoglicemiche, polidispnea e desaturazioni per cui la bambina è stata trasferita in Terapia Intensiva Neonatale (TIN) e ha necessitato di infusione con soluzione glucosata al 5%, poi sospesa, e supplementazione di O2.
All’ecocardiografia escluse cardiopatie strutturali ma visualizzati segni indiretti di ipertensione polmonare ed ipertrofia del ventricolo destro in assenza di ostruzioni all’efflusso. Il quadro è risultato compatibile con costrizione del dotto di Botallo in utero ed è nettamente migliorato dopo 10 giorni di ossigenoterapia.
Lo screening malformativo ha evidenziato unicamente rene sinistro ptosico in sede sacrale.
La neonata è stata dimessa con programma di follow-up genetico e cardiologico.
Per la presenza dei problemi clinici sopra citati, è stato eseguito presso altra sede sequenziamento dell’esoma (WES, Whole Exome Sequencing) che ha identificato la presenza della variante in eterozigosi p.Arg490ter del gene KMT2D, associata alla sindrome di Niikawa-Kuroki.
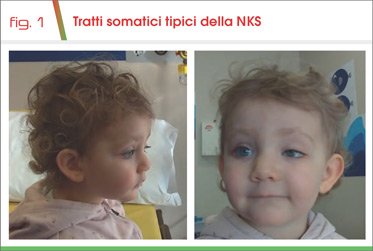 Alla nostra prima valutazione, all’età di 16 mesi, la piccola si presentava in buone condizioni generali con crescita regolare secondo le curve della popolazione generale (50° percentile) e con lieve ritardo dello sviluppo psicomotorio. Si rilevava ptosi palpebrale dell’occhio sinistro con fenomeno di Marcus-Gunn, accenno a bifidità dell’ugola e palato ogivale. Inoltre, si evidenziavano tratti compatibili con la condizione riscontrata (Fig. 1): fessure palpebrali allungate, sopracciglia arcuate e rade nel terzo laterale, punta nasale depressa e columella breve, padiglioni auricolari grandi. La problematica cardiologica risultava risolta e, dalla dimissione dalla neonatologia, non erano più state segnalate crisi ipoglicemiche.
Alla nostra prima valutazione, all’età di 16 mesi, la piccola si presentava in buone condizioni generali con crescita regolare secondo le curve della popolazione generale (50° percentile) e con lieve ritardo dello sviluppo psicomotorio. Si rilevava ptosi palpebrale dell’occhio sinistro con fenomeno di Marcus-Gunn, accenno a bifidità dell’ugola e palato ogivale. Inoltre, si evidenziavano tratti compatibili con la condizione riscontrata (Fig. 1): fessure palpebrali allungate, sopracciglia arcuate e rade nel terzo laterale, punta nasale depressa e columella breve, padiglioni auricolari grandi. La problematica cardiologica risultava risolta e, dalla dimissione dalla neonatologia, non erano più state segnalate crisi ipoglicemiche.
All’ultima valutazione, all’età di 2 anni e 5 mesi, la bambina mostrava una buona evoluzione con crescita staturale in iniziale deflessione sulle curve della popolazione generale (30° percentile) ma regolare per sindrome di Niikawa-Kuroki (50° percentile su curve specifiche) e sviluppo psicomotorio con buone acquisizioni. Gli unici problemi medici riportati erano frequenti episodi di otite media acuta e stipsi.
La sindrome di Niikawa-Kuroki
La sindrome di Niikawa-Kuroki (NKS) è una condizione genetica rara, caratterizzata da facies tipica, deficit della crescita post-natale, ritardo di sviluppo psicomotorio e/o disabilità intellettiva lieve-moderata, anomalie scheletriche minori e altre possibili anomalie congenite.
Descritta per la prima volta in Giappone, è stata osservata in tutti i gruppi etnici e si stima che abbia un’incidenza di un caso ogni 32.000 nati, senza particolari differenze di genere.
Tale condizione è nota anche come sindrome Kabuki per la somiglianza dei tratti somatici con il trucco degli attori del teatro giapponese Kabuki. Questa definizione viene sconsigliata perchè ritenuta irrispettosa verso i pazienti.
Elementi clinici di sospetto diagnostico
Il sospetto clinico si basa principalmente sulla presenza di una facies suggestiva associata a ritardo psicomotorio/disabilità intellettiva.
I dismorfismi facciali maggiormente caratterizzanti sono la presenza di sopracciglia arcuate e diradate nel terzo laterale, rime palpebrali allungate con eversione del 3° laterale della palpebra inferiore, columella breve con punta del naso depressa, padiglioni auricolari anteversi.
 Suggestivi sono anche i “fetal pads”, ovvero la presenza di cuscinetti a livello dei polpastrelli. Frequentemente riscontrate sono anche la clinodattilia del 5° dito delle mani, la brachidattilia e l’ipodontia.
Suggestivi sono anche i “fetal pads”, ovvero la presenza di cuscinetti a livello dei polpastrelli. Frequentemente riscontrate sono anche la clinodattilia del 5° dito delle mani, la brachidattilia e l’ipodontia.
La diagnosi di NKS è stabilita in un soggetto di qualsiasi età con anamnesi positiva di ipotonia infantile, ritardo di sviluppo psicomotorio e/o disabilità intellettiva e uno o più criteri riassunti in tabella 1.
Dati auxologici
Mentre i parametri auxologici in utero e alla nascita sono generalmente normali, è tipica la presenza di ritardo di crescita post-natale, di gravità variabile, con altezza finale generalmente al di sotto del 50° percentile. Rilevabile talvolta il deficit di GH.
Non costante, ma frequente, è la presenza di microcefalia. Molti pazienti sviluppano nel tempo sovrappeso o franca obesità.
Malformazioni maggiori e complicanze mediche associate
I pazienti affetti da NKS possono presentare malformazioni a livello di diversi organi ed apparati. Tra le più frequenti si rilevano quelle cardiache (coartazione dell’aorta, difetto interventricolare, difetto interatriale ecc.) e renali (ipoplasia, displasia, rene a ferro di cavallo, reflusso vescico-ureterale). Le anomalie genitali sono più frequenti nel sesso maschile e comprendono criptorchidismo, micropene ed ipospadia. Occasionalmente si rilevano alterazioni a carico del sistema nervoso centrale (malformazione di Dandy-Walker, ipoplasia del corpo calloso, anomalia di Chiari) e a livello ano-rettale (atresia anale, ano imperforato, fistola ano-vestibolare).
La NKS può essere complicata da anomalie del sistema immunitario (ipogammaglobulinemia, deficit di IgA, iper-IgM, malattie autoimmuni) con aumentata suscettibilità alle infezioni. Molto frequenti in età pediatrica le otiti medie con conseguente ipoacusia trasmissiva. Circa il 15% dei pazienti sviluppa epilessia con età di insorgenza variabile. Si rilevano occasionalmente problematiche endocrinologiche tra cui deficit di GH, telarca prematuro, diabete mellito di tipo I ed ipotiroidismo congenito. Possibile l’insorgenza di crisi ipoglicemiche soprattutto nel periodo neonatale e durante i primi mesi di vita. Da monitorare nel tempo l’insorgenza di anomalie a carico dell’apparato scheletrico, quali scoliosi (secondaria a ipotonia muscolare e/o a malformazioni vertebrali) e piattismo dei piedi. In circa il 75% dei pazienti è presente iperlassità legamentosa che può causare lussazione congenita delle anche e lussazione della rotula.
Sviluppo psico-intellettivo e fenotipo comportamentale
I bambini affetti da NKS presentano generalmente ritardo psicomotorio di entità lieve-moderata, con particolare difficoltà nell’espressione verbale. Sono in genere descritti come estroversi e socievoli. È raramente possibile osservare un quadro di disturbo dello spettro autistico.
La maggior parte dei bambini frequenta la scuola con insegnante di sostegno e programma didattico personalizzato. Gli adulti acquisiscono solitamente competenze tali da poter svolgere lavori semplici in ambienti protetti anche se difficilmente raggiungono l’autosufficienza completa.
Difetto genetico di base
La NKS fa parte del gruppo delle cromatinopatie, condizioni causate da alterazione dei meccanismi di regolazione epigenetica della cromatina.
Ad oggi nell’80% circa delle persone con diagnosi clinica viene identificato il difetto genetico causativo. Il 75% dei pazienti presenta una variante patogenetica a carico del gene KMT2D in eterozigosi (12q13.12). Nel 3-5% dei pazienti vengono invece osservate anomalie nel gene KDM6A (Xp11.3).
Nella maggior parte dei casi si tratta di varianti de novo; solo in una piccola percentuale di casi la mutazione viene trasmessa come carattere autosomico dominante (KDT2D) o X-linked recessivo (KDM6A).
Trattamento
I bambini affetti da NKS necessitano di una presa in carico multidisciplinare, volta al monitoraggio clinico e alla prevenzione delle possibili complicanze mediche descritte.
Decisivo è il percorso riabilitativo che deve necessariamente includere una particolare attenzione alla promozione della comunicazione anche attraverso approcci non verbali (Comunicazione Aumentativa Alternativa).
Associazione di genitori
Dal 2015 è attiva AISK, l’Associazione Italiana Sindrome Kabuki www.sindromekabuki.it che riunisce i pazienti affetti da questa malattia genetica rara e le loro famiglie.
Bibliografia
- Wang YR, Xu NX, Wang J, Wang XM. Kabuki syndrome: review of the clinical features, diagnosis and epigenetic mechanisms. World J Pediatr. 2019;15:528–535.
- Selicorni A, Zampino G, Memo L, Scarano G. Sindromi malformative: una guida per il pediatra. 2017. Pacini Editore Medicina.
- Boniel S, Szymańska K, Śmigiel R, Szczałuba K. Kabuki Syndrome - Clinical Review with Molecular Aspects. Genes (Basel). 2021; 12(4):468.
- Adam MP, Banka D, Bjornsson HT, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019; 56(2):89-95.