




la Newsletter

Erika Apuril1,2, Milena Mariani1
1UOC Pediatria. Presidio S. Fermo. ASST-Lariana, Como. Centro Fondazione Mariani per il Bambino Fragile; 2Università degli Studi di Milano - Scuola di Specializzazione in Genetica Medica
La sindrome di Pallister Killian | Facies grossolana con profilo...
Facies grossolana con profilo piatto, calvizie frontotemporale,...
Presentiamo il caso di una piccola paziente giunta in visita presso l'ambulatorio di Genetica Pediatrica.
Terzogenita, di genitori sani non consanguinei, assenza di familiarità per patologie di rilevanza genetica. Diabete gestazionale risoltosi con insulinoterapia. I controlli ecografici erano nei limiti di norma. Nata da parto eutocico a 38 sg, peso 3750 kg, lunghezza 51 cm, CC 34 cm, APGAR 9/10. Alla nascita riscontro di dismorfismi ed ano anteriorizzato. Allo screening malformativo vengono evidenziate stenosi sopravalvolare polmonare e iperecogenicità periventricolare anteriore sinistra del quarto ventricolo. Alla visita neuropsichiatrica (NPI) viene segnalato repertorio motorio povero.
Il follow-up ha mostrato la presenza di un modesto ritardo psicomotorio con buona evoluzione. A 17 mesi di vita, riscontro di lesioni iperpigmentate al tronco e lungo gli arti inferiori con aspetto lineare e spiraliforme lungo le linee di Blaschko; impianto notevolmente arretrato dei capelli in regione temporo-occipitale bilateralmente. Il riscontro di questi dati ha suggerito l’esecuzione di un cariotipo su biopsia cutanea che ha mostrato la presenza di due linee cellulari: un cariotipo femminile normale (3 colonie) e l’altra a 47 cromosomi con riscontro di isocromosoma 12p (22 colonie). Tale reperto ha permesso di confermare il sospetto clinico di sindrome di Pallister-Killian.
Successivamente alla diagnosi riscontro di ipoacusia mista di grado medio, astigmatismo ipermetropico, e corpo calloso assottigliato alla RMN encefalo.
La piccola ha oggi 8 anni e 7 mesi, la sua crescita è regolare con tendenza al sovrappeso (97°p) e porta protesi uditive. Esegue periodicamente controlli specialistici, tra cui visita oculistica, cardiologica e NPI (ad oggi EEG nella norma). Pratica nuoto, psicomotricità, logopedia e Comunicazione Aumentativa Alternativa (CAA) con grande beneficio.
La sindrome di Pallister Killian
La sindrome di Pallister Killian (PKS) è una rara anomalia cromosomica a insorgenza sporadica caratterizzata da tetrasomia a mosaico del braccio corto del cromosoma 12 (tetrasomia 12p). La reale prevalenza di questa malattia è incerta, stimata intorno a 1/20.000-25.000 nati vivi/anno.
L’eccesso di materiale genetico è responsabile delle caratteristiche distintive della PKS: tratti peculiari del volto (fronte ampia ed alta, calvizie fronto-temporale, ciglia e sopracciglia rade, ipertelorismo, upslanting, radice nasale piatta e allargata, naso corto con narici rivolte verso l’alto, padiglioni auricolari leggermente retro ruotati a basso impianto, filtro lungo a “V” prominente, bocca larga con angoli rivolti verso il basso e labbro superiore sottile, macroglossia, micrognatia) e ritardo psicomotorio associati nel 50% dei casi a epilessia.
Elementi clinici di sospetto diagnostico
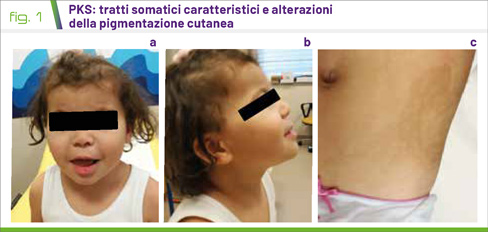
Alla nascita la presenza di ipotonia associata a tratti peculiari del viso rappresenta il campanello d’allarme più specifico; nel corso degli anni i tratti del volto diventano più grossolani (Fig. 1 a e b). Il caratteristico impianto dei capelli tende ad essere meno evidente con il passare degli anni. Spesso possono essere evidenziate anomalie della pigmentazione cutanea espressione della distribuzione a mosaico della tetrasomia (Fig. 1 c).
Dati auxologici
I parametri auxologici tendono ad essere nella norma/ai limiti superiori della norma rispetto alla popolazione generale con rallentamento dell’incremento ponderale nel primo anno di vita associato alle problematiche alimentari. Viene descritta obesità nell’età adolescenziale/adulta.
Sviluppo psico-intellettivo e fenotipo comportamentale
Il ritardo psicomotorio è solitamente di grado moderato-severo anche se sono stati descritti più raramente bambini con un ritardo lieve. A livello comportamentale sono descritte stereotipie, auto-aggressività, anomalie del ritmo sonno-veglia.
Malformazioni maggiori e complicanze mediche associate
Sono descritti difetti congeniti a carico del cuore, del sistema nervoso centrale, del sistema genito-urinario e del diaframma; nessuna malformazione è però patognomonica della condizione. Possono essere frequenti problematiche respiratorie, reflusso gastroesofageo, stipsi, e difficoltà alimentari secondarie all’ipotonia. È descritta l’insorgenza di epilessia nei primi anni di vita, raramente in epoca neonatale; le crisi possono avere una semeiotica variabile; non è descritto un pattern particolare all’EEG. Frequenti sono inoltre ipoacusia neurosensoriale, problematiche visive, disfunzione autonomica (anidrosi/ipoidrosi), iperventilazione ed altre anomalie del pattern respiratorio.
Difetto genetico di base
L’esame gold standard per la diagnosi è rappresentato dal cariotipo su un tessuto differente dai linfociti del sangue periferico (es. fibroblasti da biopsia cutanea) in quanto l’indagine condotta sui linfociti, seppur meno invasiva, può essere spesso negativa perché la percentuale di isocromosoma 12p in queste cellule si attesta in genere attorno allo 0-2%.
Ad oggi vi sono però evidenze relative alla capacità dell’arrayCGH di rilevare l’anomalia anche su sangue periferico con il vantaggio di una minore invasività rispetto alla biopsia cutanea.
I dati disponibili segnalano l’assenza di correlazione tra entità del mosaicismo e gravità/benignità del fenotipo clinico.
Trattamento
Non esiste una terapia specifica per le persone affette da PKS: è indispensabile attivare un percorso riabilitativo per potenziare le performance neurocognitive. Va inoltre impostato un monitoraggio clinico mirato volto alla prevenzione/trattamento delle possibili complicanze mediche associate.
PKS Italia Aps è l'Associazione Italiana dedicata alla Sindrome di Pallister-Killian:
www.pksitalia.org - info@pksitalia.org
Bibliografia
- Stephens CM, Pavel AM, Mathieson SR, et al. Case Report: Early Neonatal EEG in Two Infants with Pallister Killian Syndrome (PKS). HRB Open Res. 2022;5:14.
- Fetta A, Soliani L, Trevisan A, et al. Cognitive, Behavioral, and Sensory Profile of Pallister-Killian Syndrome: A Prospective Study of 22 Individuals. Genes (Basel). 2022;13(2):356.
- Gigliotti MJ, Tachie-Baffour Y, Jafrani RJ, et al. A Novel Case of Tethered Cord in a Five-Month-Old Male With Pallister-Killian Syndrome. Cureus. 2020;12:e11240.
- Salzano E, Raible SE, Kaur M, et al. Prenatal profile of Pallister-Killian syndrome: Retrospective analysis of 114 pregnancies, literature review and approach to prenatal diagnosis. Am J Med Genet A. 2018;176(12):2575-2586.