




la Newsletter

Massimo Maschio, Beatrice Belluzzi
Centro Regionale per la diagnosi e la cura della Fibrosi Cistica, Clinica Pediatrica - IRCCS Burlo Garofolo, Trieste
La sindrome pseudo-Bartter nella fibrosi cistica | Nei bambini con...
Nei bambini con fibrosi cistica che presentano segni clinici di...
Riportiamo il caso di un lattante di 9 mesi, affetto da fibrosi cistica (FC), omozigote per la mutazione delta F508, che in corso di controllo periodico presenta mancata crescita con calo ponderale (peso 7810 g, ridotto di 120 g rispetto a tre settimane prima), e riscontro all’emogasanalisi (EGA) capillare di alcalosi metabolica (pH 7.54, pCO2 40.6, HCO3- 34,8). La sua storia precedente era stata caratterizzata da scarsa crescita persistente, nonostante adeguato dosaggio di Creon, ed erano già stati necessari tre ricoveri presso la nostra Clinica Pediatrica per tre episodi di disidratazione severa associata ad alcalosi metabolica.
In occasione del primo ricovero a 3 mesi di vita aveva presentato calo ponderale, segni clinici di disidratazione (alonamento perioculare, cute disidratata, iporeattività) e all’EGA alcalosi metabolica iponatriemica ipocloremica (pH 7.57, HCO3- 46.5 mmol/L con Na+ 125 mEq/L, Cl- 64 mEq/L). Pertanto, era stata avviata idratazione endovenosa con progressivo aumento ponderale e ripristino della normalizzazione degli elettroliti e dell’EGA.
Durante i successivi ricoveri, avvenuti a 4 e a 6 mesi di vita, per il persistere della scarsa crescita nei mesi intercorrenti, era stato posizionato un Sondino Naso-Gastrico (SNG) per consentire il mantenimento di una reidratazione anche a domicilio, che, tuttavia, il bambino si era sfilato pochi giorni dopo il ricovero, e che non era più stato riposizionato per il contestuale avvio dell’alimentazione complementare.
Considerati i precedenti ricoveri ed il nuovo riscontro di alcalosi metabolica e calo ponderale, si è deciso di posizionare nuovamente il SNG per avviare l’idratazione e, inoltre, di valutare la necessità di gastrostomia percutanea (PEG). Nell’ultimo periodo, infatti, nonostante l’indicazione a supplementare l’apporto idrosalino per bocca con la soluzione reidratante orale (SRO) nell’arco della giornata, il bambino tendeva a rifiutarne l’assunzione (massimo 50-100 mL/die), e si presentava un contesto familiare di inadeguata gestione dell’apporto nutrizionale legato, in particolare, a barriere linguistiche importanti.
Dunque, per l’elevato rischio di disidratazione, è stato proposto alla famiglia il posizionamento di una PEG attraverso cui supplementare i liquidi, difficilmente assunti per bocca, considerata la scarsa tolleranza dimostrata in precedenza verso il SNG.
Con l’avvio della reidratazione via PEG il bambino non ha più presentato episodi di disidratazione severa né ha avuto necessità di ricovero, riportando, inoltre, uno sviluppo ponderale stabile e adeguato secondo i percentili di crescita per età.
Patogenesi
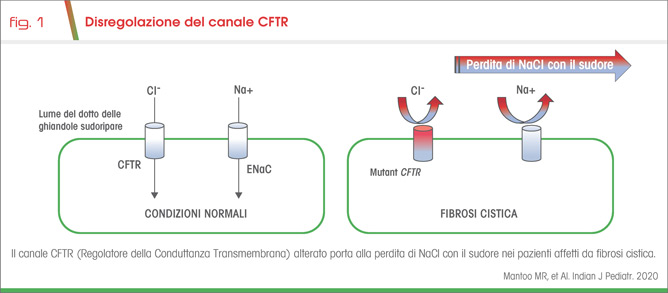
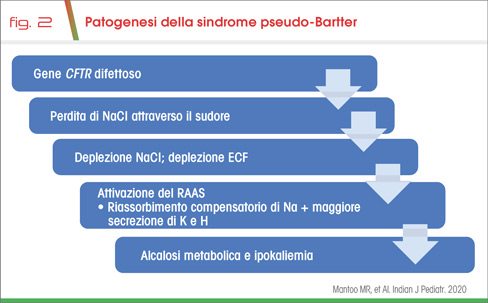
La fibrosi cistica (FC) è dovuta alla mutazione del gene CFTR e comporta la disregolazione dell’omonimo canale, che ha la funzione di regolare il trasporto di cloro e bicarbonati attraverso la membrana apicale di diversi epiteli quali, ad esempio, quello delle ghiandole sudoripare (1,2). La sua anormale attività a tale livello favorisce un ridotto assorbimento e dunque una perdita di NaCl con il sudore (Fig 1) (2), che, qualora eccessiva, comporta iponatremia ed ipocloremia. Esse si associano ad alcalosi metabolica ed ipokaliemia per l’attivazione del sistema RAAS, che provoca l’aumento dell’aldosterone e dunque una maggior secrezione di K+ e H+ dal dotto collettore renale (Fig 2) (2).
Diagnosi
 Tra i possibili sintomi di presentazione della FC è importante considerare anche le alterazioni elettrolitiche e dell’equilibrio acido-base: alcalosi metabolica ipocloremica, iponatriemica ed ipokaliemica (2-4).
Tra i possibili sintomi di presentazione della FC è importante considerare anche le alterazioni elettrolitiche e dell’equilibrio acido-base: alcalosi metabolica ipocloremica, iponatriemica ed ipokaliemica (2-4).
Queste caratteristiche prendono il nome di sindrome pseudo-Bartter e clinicamente possono manifestarsi in modo subacuto o cronico, con disidratazione, sudorazione eccessiva, febbre, infezioni respiratorie, vomito e scarsa crescita.
Esse sono favorite da condizioni di maggiore sudorazione (temperature elevate, esercizio fisico) e sono più frequenti sotto l’anno di vita per la scarsa supplementazione di NaCl derivante dall’assunzione di latte materno (< 7 mmol/l) o di formula (< 15 mmol/L) (2,3). I sintomi diselettrolitici, se sospettati, vanno tempestivamente valutati attraverso il dosaggio degli elettroliti ematici e l’esecuzione di un EGA.
Diagnosi differenziale
La sindrome pseudo-Bartter può associarsi anche ad altre condizioni, legate ad una perdita di NaCl per via:
- urinaria: sovradosaggio di diuretico dell’ansa (furosemide), nefrotossicità da farmaci (amfotericina B)
- gastrointestinale: stenosi ipertrofica del piloro (vomiti ripetuti), abuso di lassativi, cloridorrea congenita (2).
Inoltre, è importante distinguerla dalla classica sindrome di Bartter, dovuta all’alterazione del riassorbimento del NaCl a livello del tratto ascendente dell’ansa di Henle renale (2). Esse possono essere distinte valutando la frazione di escrezione di Na+ urinario (FeNa+): bassa nella pseudo-Bartter, elevata nella Bartter (5).
Terapia e prevenzione
Il trattamento della sindrome pseudo-Bartter prevede il reintegro immediato di liquidi e sali attraverso la via endovenosa. Tuttavia, la prevenzione della disidratazione, soprattutto nei mesi estivi o in corso di vomito e/o diarrea profusi, risulta fondamentale (2,7). È raccomandato supplementare il NaCl in tutti i bambini affetti da FC, inizialmente al dosaggio di 1-2 mmol/kg/die (max 4 mmol/kg/die) con monitoraggio dell’escrezione urinaria di Na+ che va mantenuta tra lo 0.5-1.5%. Nei bambini più grandi è possibile incrementare l’assunzione di cibi salati o somministrare direttamente compresse di NaCl o la stessa soluzione reidratante orale (SRO) (6). Nel caso in cui non sia possibile garantire una supplementazione adeguata per via orale, è opportuno valutare la necessità di una nutrizione enterale, in prima battuta tramite SNG e, se mal tollerato, tramite PEG, come nel nostro caso.
Conclusioni
Nei bambini affetti da FC che presentano segni clinici di disidratazione o una storia persistente di scarsa crescita, nonostante adeguato dosaggio di Creon, è opportuno sospettare una sindrome da perdita di sali, soprattutto se di età inferiore all’anno di vita e nei mesi estivi. Inoltre, va prontamente valutata la necessità di avvalersi della nutrizione enterale, per garantire la giusta reintegrazione di NaCl e scongiurare un quadro di alcalosi metabolica e diselettrolitico persistenti.
Bibliografia
- De Boeck K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020;109(5):893-899.
- Mantoo MR, et al. Cystic Fibrosis Presenting as Pseudo-Bartter Syndrome: An Important Diagnosis that is Missed! Indian J Pediatr. 2020;87(9):726-732.
- Scurati-Manzoni E, et al. Electrolyte abnormalities in cystic fibrosis: systematic review of the literature. Pediatr Nephrol. 2014;29(6):1015-23.
- Faraji-Goodarzi M. Pseudo-Bartter syndrome in children with cystic fibrosis. Clin Case Rep. 2019;7(6):1123-1126.
- N. Della Vecchia, M. Mariani. Sindrome pseudo-Bartter e disidratazione: presentazione atipica di fibrosi cistica. Medico e Bambino pagine elettroniche 2019;22(7):153 https://www.medicoebambino.com/?id=PSR1908_20.html
- Turck D, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. 2016;35(3):557-77.
- Botti M, et al. Sindrome da perdita di sali e infezione da Clostridium difficile nella fibrosi cistica. Medico e Bambino 2022;25(1): e21-e22.