




la Newsletter

Paolo Manzi1, Michelina Sibilio2, Anita Vergatti1, Nadia Altavilla1, Domenico Rendina1, Antonio Barbato1
1Dipartimento di Medicina Clinica e Chirurgia, Università degli Studi di Napoli Federico II, Napoli; 2UOSD di Malattie Metaboliche, AORN Santobono-Pausilipon Ospedale SS. Annunziata, Napoli
La transizione dalla pediatria alla medicina dell’adulto: un...
Il trattamento precoce con la ERT può rallentare la progressione...
Nel 2006 S.A., un bambino di 5 anni, giungeva presso il Dipartimento di Pediatria dell’AOU Federico II per ipercifosi lombare con tendenza alla scoliosi e varismo della tibia comparsi all’età di 20 mesi.
Lo screening metabolico evidenziava:
- elevati livelli urinari di glicosaminoglicani (GAGs) (158,4 mgGAG/g creatinina; v.n.: 22-88)
- assenza dell’attività enzimatica della N-acetilgalattosammina-6 solfatasi (GALNS)
- presenza delle mutazioni [c.274C>T (p.Pro92Ser)] e [c.895C>T (p.Gln299*)] nel gene GALNS (1,2).
Sulla base del quadro clinico, laboratoristico e molecolare veniva posta diagnosi di mucopolisaccaridosi tipo IV A (MPS IV A o sindrome di Morquio).
Per le problematiche ortopediche, dall’età di 7 anni il paziente è stato seguito presso l’Istituto Ortopedico Rizzoli di Bologna. A 10 anni veniva sottoposto ad intervento di allungamento growing rod vertebrale con successivi interventi annuali di rimodellamento fino all’età di 16 anni. A 18 anni si sottoponeva a intervento di sostituzione protesica dell’anca destra e a 21 anni dell’anca sinistra.
A 14 anni iniziava la terapia enzimatica sostitutiva (ERT) per via endovenosa con elosulfase alfa alla dose di 2 mg/kg ogni settimana con buona compliance terapeutica ed in assenza di reazioni avverse. Prima dell’inizio della ERT venivano praticati i seguenti esami:
- RMN encefalo: evidenziava dismorfismi a carico delle vertebre cervicali con impressione basilare con platibasia, ed ispessimento dei tessuti legamentosi e durali peri-odonotodei (3) e dislocazione caudale delle tonsille cerebellari attraverso il forame occipitale e ridotti spazi liquorali a livello della cisterna magna e del forame occipitale, associati a malformazioni di Chiari 1 (4).
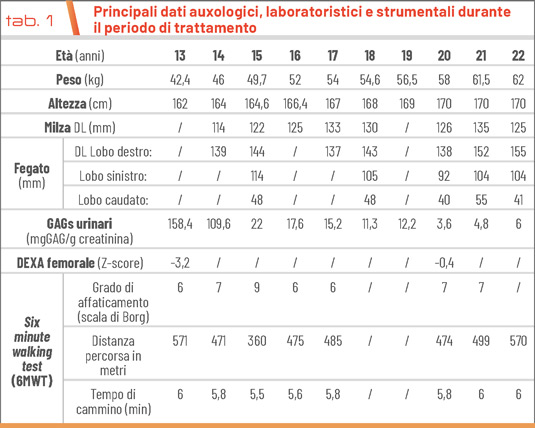
- Ecografia addome: mostrava lieve epatomegalia (DL lobo destro: 139 mm) e lieve splenomegalia (DL: 114 mm) (Tab. 1).
- Ecocardiografia: nella norma.
 A 18 anni transitava, dopo visita collegiale con i pediatri, alla Medicina Interna dell’AOU Federico II presso l’ambulatorio di Malattie d’accumulo lisosomiale dell’adulto per il prosieguo del follow-up.
A 18 anni transitava, dopo visita collegiale con i pediatri, alla Medicina Interna dell’AOU Federico II presso l’ambulatorio di Malattie d’accumulo lisosomiale dell’adulto per il prosieguo del follow-up.
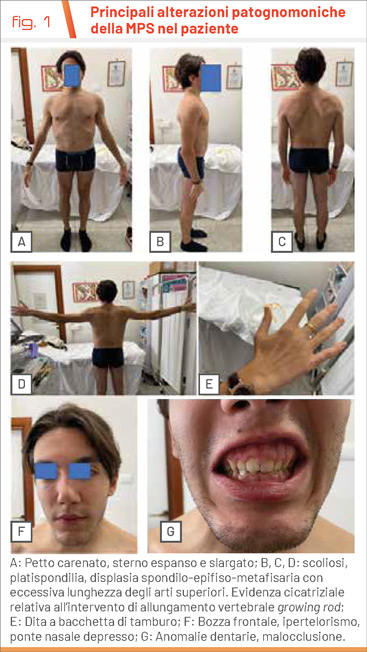
Dopo 8 anni di ERT, gli esami ematochimici non hanno evidenziato alterazioni della funzionalità epatica, renale e nutrizionale. L’ecocardiografia ha mostrato insufficienza mitralica, tricuspidalica e polmonare di grado lieve, in assenza d’ispessimento valvolare e con una normale frazione d’eiezione (62%; v.n.: >53). I GAGs urinari risultavano nella norma (6 mgGAG/g creatinina; v.n.: 3-30). La valutazione della densità ossea a livello del collo del femore valutata mediante densitometria ossea (DEXA) ha evidenziato un netto miglioramento dopo 7 anni di trattamento (T0: 0.6 g/cm2 e Z-score: -3.1; dopo 7 anni di trattamento: 1.021 g/cm2 e Z-score: -0.4). Per la comparsa di valori di pressione arteriosa elevati confermati in diverse occasioni, è stato introdotto in terapia ramipril 5 mg/die con normalizzazione dei livelli pressori. Durante l’ultima visita, il paziente ha evidenziato la sua volontà di sospendere la ERT sia per la frequenza delle somministrazioni, poco compatibili con lo stile di vita di uno studente universitario, che per le difficoltà incontrate presso il Centro infusore. Dopo un’accurata valutazione del rapporto costo benefici, alla luce dei risultati ottenuti in questi anni, almeno in parte imputabili alla ERT, il paziente ha deciso per il momento di continuare la terapia. In tabella 1 sono riportati i principali parametri nella fase di transizione dall’età pediatrica a quella adulta. La figura 1 mostra le principali alterazioni patognomoniche della MPS nel paziente.
Patogenesi e manifestazioni cliniche
 Le mucopolisaccaridosi (MPS) costituiscono un gruppo di malattie rare da accumulo (incidenza: 1:25.000 nati vivi) dovute al deficit di specifici enzimi lisosomiali che degradano i GAGs. Il deficit enzimatico determina, quindi, l’accumulo intracellulare di GAGs nei tessuti e nei fluidi biologici, in particolare sangue e urine. Una diagnosi precoce è necessaria per ottimizzare il trattamento, prevenendo così danni irreversibili a tessuti e organi (5), in particolare per quelle forme (come la IV A) per cui sono disponibili approcci terapeutici quali la ERT (6).
Le mucopolisaccaridosi (MPS) costituiscono un gruppo di malattie rare da accumulo (incidenza: 1:25.000 nati vivi) dovute al deficit di specifici enzimi lisosomiali che degradano i GAGs. Il deficit enzimatico determina, quindi, l’accumulo intracellulare di GAGs nei tessuti e nei fluidi biologici, in particolare sangue e urine. Una diagnosi precoce è necessaria per ottimizzare il trattamento, prevenendo così danni irreversibili a tessuti e organi (5), in particolare per quelle forme (come la IV A) per cui sono disponibili approcci terapeutici quali la ERT (6).
Mucopolisaccaridosi IV A
La MPS IV A è causata dal deficit dell’enzima GALNS, responsabile della degradazione del cheratan solfato (KS), trasmessa con modalità autosomica recessiva. Il gene responsabile (OMIM # 253000) è localizzato sul cromosoma 16 (1). La prevalenza è circa 1:250.000 nati vivi. La MPS IV A è caratterizzata da importanti alterazioni scheletriche che diventano man mano più evidenti con la crescita del bambino. Il coinvolgimento scheletrico causa il deterioramento della deambulazione e delle attività quotidiane, a seconda della gravità della malattia. I sintomi extra-scheletrici possono comprendere problemi respiratori con patologia polmonare restrittiva, epatomegalia, splenomegalia, valvulopatia e coinvolgimento cardiaco, sordità e opacità corneali. L’intelligenza è normale, la facies non sempre è patognomonica. Verso i 5-6 anni di età l’ipoplasia del processo odontoideo e l’iperlassità articolare, qualora presenti, possono causare instabilità delle prime due vertebre cervicali. è descritto un aumentato rischio anestesiologico.
Diagnosi
Nel bambino il coinvolgimento osseo rappresenta il quadro clinico più frequente ad indirizzare il sospetto diagnostico (2). Il dosaggio quantitativo e qualitativo dei GAGs urinari (il cheratan solfato risulta elevato nel caso specifico della MPS IV A) è il test di primo livello in caso di sospetto clinico, ma la diagnosi di conferma deve essere effettuata mediante il dosaggio dell’attività enzimatica e all’analisi molecolare del gene GALNS (1). La diagnosi prenatale è possibile mediante villocentesi o amniocentesi nelle coppie a rischio (5).
Trattamento
Il trattamento con elosulfase alfa rappresenta un approccio terapeutico efficace per il trattamento di alcune manifestazioni della MPS IV A. Un trattamento precoce della patologia con la ERT può rallentare la progressione della malattia, migliorare la funzione polmonare e la qualità di vita dei pazienti (6). In letteratura rari sono i dati disponibili che documentano l’efficacia della ERT sull’aspetto osseo, tuttavia nel paziente da noi descritto la DEXA femorale ha evidenziato un netto miglioramento della densità ossea dopo 7 anni di trattamento. Nel caso clinico descritto, in accordo a quanto riportato in letteratura, abbiamo osservato la normalizzazione dei livelli di GAGs urinari, la stabilità della volumetria degli organi ipocondriaci e la buona tolleranza allo sforzo fisico dimostrata con il test del cammino dei 6 minuti (Tab. 1).
Conclusioni
Per la gestione di un paziente affetto da MPS IV A è fondamentale un approccio multidisciplinare a causa delle molteplici complicanze che questi pazienti possono sviluppare nel tempo. La transizione dalla pediatria alla medicina dell’adulto deve essere quindi un continuum dove un case manager, quale il medico internista, è in grado di coadiuvare i vari specialisti coinvolti nel follow-up nell’ottica di un approfondimento delle conoscenze sul decorso della malattia mantenendo l’efficacia dei trattamenti a lungo termine.
Bibliografia
- Longo N, Dimmock D, Levy H, et al. Evidence- and consensus-based recommendations for the use of pegvaliase in adults with 1. Za1. Zanetti A, D'Avanzo F, AlSayed M, et al. Molecular basis of mucopolysaccharidosis IVA (Morquio A syndrome): A review and classification of GALNS gene variants and reporting of 68 novel variants. Hum Mutat. 2021; 42(11):1384–1398.
- Sawamoto K, Álvarez González JV, Piechnik M, et al. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int J Mol Sci. 2020;21(4):1517.
- Padash S, Obaid H, Henderson RDE, et al. A pictorial review of the radiographic skeletal findings in Morquio syndrome (mucopolysaccharidosis type IV). Pediatr Radiol. 2023;53(5):971-983.
- Shapiro EG, Eisengart JB. The natural history of neurocognition in MPS disorders: A review. Mol Genet Metab. 2021;133(1):8-34.
- Donati MA, Pasquini E, Spada M, et al. Newborn screening in mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):126.
- Lee CL, Chuang CK, Chiu HC, et al. Clinical Utility of Elosulfase Alfa in the Treatment of Morquio A Syndrome. Drug Des Devel Ther. 2022;16:143-154.