




la Newsletter
L’atresia congenita digiuno-ileale complessa: nuove possibilità...
L’atresia digiuno-ileale è tra le più comuni anomalie...
Definizione, incidenza ed epidemiologia
L’intestino tenue nel suo tratto libero nella cavità peritoneale, fra il legamento di Treitz e la valvola ileo-ciecale, durante la fase dell’organogenesi può subire anomalie e alterazioni di calibro in forma di atresia o di stenosi.
Per atresia congenita digiuno-ileale si intende la completa ostruzione del lume del piccolo intestino in uno o più tratti. Le ostruzioni incomplete o parziali - stenosi intestinali - riconoscono gli stessi fenomeni causali delle atresie, sono molto meno frequenti e fenotipicamente danno origine a manifestazioni cliniche meglio tollerate e più tardive.
L’atresia rappresenta la quasi totalità di questi casi, ed ha una incidenza di circa 1:1500 nati vivi, senza sostanziale differenza fra i sessi.
L’atresia digiuno-ileale è quindi tra le più comuni anomalie congenite del piccolo intestino e la causa principale di ostruzione intestinale nei neonati.
La prematurità è riconosciuta come un fattore di rischio per lo sviluppo di atresia digiuno-ileale.
Si definisce pretermine un neonato che nasca prima della 37ma settimana di età gestazionale. Le cause di nascita pretermine sono numerose e complesse, la sua fisiopatologia è per lo più sconosciuta, ma è noto il coinvolgimento di fattori predisponenti materni, fetali e placentari. Tra questi i principali includono: emorragia ante-partum, fattori meccanici correlati all’utero, alterazioni ormonali, infezioni batteriche e stati infiammatori.
Anche il basso peso alla nascita costituisce un fattore di rischio importante: un terzo dei pazienti con atresia digiunale, un quarto di quelli con atresia ileale e più della metà di quelli con atresia multipla presentano un peso alla nascita nettamente inferiore alla media.
Cenni storici
Goeller nell’anno 1683 fu probabilmente il primo a fornire una descrizione di atresia ileale. Altri casi sono stati riportati rispettivamente nel 1773 e nel 1779 da Calder e da Osiander. Nei primi anni del 1800, Voisin confezionò una enterostomia in un’atresia ileale e Meckel ne pubblicò una revisione accennando a possibili fattori eziologici della malformazione. Nel 1889 Bland-Sutton propose una classificazione dei vari tipi di atresia e avanzò l’ipotesi che tali difetti potessero avvenire in sede di eventi obliterativi embriologici come ad esempio un’atrofia del dotto vitellino. Nel 1894 Wanitschek tentò, con esito infausto, la prima resezione e anastomosi per atresia intestinale. Nel 1902 Tandler propose la prima teoria eziopatogenetica. Il primo caso trattato con successo mediante anastomosi è quello descritto da Fockens nel 1911.
Genetica
I casi di atresia digiuno-ileale sono solitamente unici nella famiglia. Sono state tuttavia descritte alcune famiglie con più soggetti colpiti da atresia del digiuno o dell’ileo, come riportato ad esempio nella prima segnalazione di Mishalany e Najjar con 4 individui affetti in una fratria di diciotto (1). In tali casi, la presenza di due o più fratelli o sorelle colpiti da atresia, figli di genitori sani spesso consanguinei, supporta l’ipotesi di una trasmissione di tipo autosomico recessivo (2), con rischio riproduttivo per i genitori pari al 25% ad ogni concepimento. I casi familiari descritti sono clinicamente eterogenei, ma corrispondono prevalentemente ai tipi IIIb (apple-peel syndrome) e IV della classificazione di Grosfeld (3).
Tra le forme familiari, esiste una rara condizione di atresia multipla che può interessare ogni punto del tratto gastrointestinale, definita atresia intestinale multipla ereditaria (Hereditary Multiple Intestinal Atresia - HMIA). Si tratta di una patologia genetica a trasmissione autosomica recessiva con maggior prevalenza in popolazioni di origine franco-canadese, che determina lesioni intestinali atresiche multiple con calcificazioni omogenee intraluminali in associazione con un’immunodeficienza combinata. Il decesso si verifica nella maggior parte dei pazienti entro i due anni di età. La patologia è causata da mutazioni bi-alleliche nel gene TTC7A (Tetratricopeptide Repeat Domain 7A) che codifica per una proteina espressa nelle cellule epiteliali del timo e dell’intestino (4). Il deficit di TTC7A sembra determinare un aumento dell’attività delle Rho-chinasi, con conseguente alterazione della polarità, della crescita e della differenziazione delle cellule epiteliali intestinali ed alterazione dell’omeostasi delle cellule immunitarie (5).
Altra condizione sindromica rara caratterizzata da atresia digiuno-ileale è la sindrome di Strømme: gli individui affetti presentano atresia intestinale di tipo IIIb, anomalie oculari, microcefalia e talvolta il coinvolgimento di altri apparati (renale, cardiovascolare). In alcuni casi la condizione è letale in epoca precoce, mentre altri pazienti presentano una normale sopravvivenza, con o senza disabilità intellettiva. La trasmissione è anche in questo caso autosomica recessiva e la patologia è stata associata a mutazioni di CENPF, gene centrosomico già implicato in altre ciliopatie fetali (6).
Ad eccezione delle rare forme sindromiche citate, l’atresia digiuno-ileale tende a presentarsi come malformazione isolata, se non quando secondaria ad altro difetto (es. gastroschisi), e si riscontrano ulteriori anomalie congenite solo in una piccola percentuale di casi a carico dell’apparato gastrointestinale, del distretto genitourinario, del cuore o del sistema nervoso centrale (7). Manifestazioni extra-intestinali tendono ad essere più frequenti nelle atresie digiunali piuttosto che nelle atresie ileali.
È inoltre noto che il 9-11% dei soggetti con atresia digiuno-ileale è affetto da fibrosi cistica. Il meccanismo di insorgenza dell’atresia è legato all’ileo da meconio. In tutti i casi di riscontro prenatale e postnatale di atresia digiuno-ileale è pertanto necessario prendere in considerazione l’ipotesi diagnostica di fibrosi cistica ed avviare il bambino ad approfondimenti diagnostici sul gene CFTR ed eventuale consulenza genetica per la famiglia (7-8). Tale associazione ha infatti rilevanti implicazioni diagnostiche e prognostiche.
Patogenesi e classificazione
La prima teoria eziopatogenetica proposta da Tandler (1902) sulla genesi delle atresie digiuno-ileali sostiene, quale evento patogenetico caratteristico, la mancata ricanalizzazione del lume intestinale al termine dello stato “solido” dello sviluppo dell’organo.
Nel corso delle prime settimane dell’embriogenesi, il lume intestinale diviene progressivamente più piccolo e temporaneamente si oblitera per la proliferazione delle sue cellule epiteliali. Normalmente avviene una vacuolizzazione per la degenerazione delle cellule epiteliali; ne consegue che l’intestino regolarmente torna ad essere canalizzato alla fine del periodo embrionale, verso la 12ma settimana di gestazione. Dalla mancata formazione di un numero adeguato di vacuoli durante la ricanalizzazione dell’intestino possono derivare anomalie determinanti un’occlusione parziale (stenosi) o completa (atresia) del lume intestinale.
In seguito hanno avuto un ruolo sempre più importante le teorie che riconoscono quale genesi di un’atresia digiuno-ileale, un insulto ischemico intrauterino dell’intestino medio con coinvolgimento di singoli o multipli segmenti dell’organo già pienamente sviluppato. L’interruzione del flusso vascolare ne determina la necrosi ischemica e asettica con successivo riassorbimento del segmento o dei segmenti coinvolti.
Uno studio condotto da Komuro e collaboratori ha chiaramente dimostrato l’associazione tra anomalie vascolari della placenta e la presenza di atresie digiuno-ileali complesse quali atresie multiple o atresie apple-peel.
 Fra le varie classificazioni proposte, la più seguita è quella di Grosfeld (9), in cui si distinguono quattro tipi di atresia digiuno-ileale di cui il tipo III è ulteriormente distinto in due sottotipi (Fig. 1):
Fra le varie classificazioni proposte, la più seguita è quella di Grosfeld (9), in cui si distinguono quattro tipi di atresia digiuno-ileale di cui il tipo III è ulteriormente distinto in due sottotipi (Fig. 1):
TIPO I (23%) detta anche atresia membranosa (diaframma): ostruzione secondaria alla presenza di una membrana o di un diaframma mucoso completo con parete intestinale intatta e mesentere integro. L’intestino prossimale è dilatato, quello distale è collabito. Con l’aumento della pressione intraluminale nella porzione prossimale si può verificare uno sfiancamento del diaframma all’interno dell’intestino distale, che viene a sua volta disteso, creando un effetto cosiddetto a “manica a vento”.
Nell’atresia di tipo I non vi è riduzione della lunghezza intestinale.
TIPO II (10%): l’atresia si estrinseca come un cordone fibroso che unisce le due tasche intestinali a monte e a valle, senza interruzione del mesentere. L’aumento della pressione al livello del moncone intestinale prossimale, che si presenta dilatato e ipertrofico, può aggravarne le condizioni determinando ischemia focale. Il moncone distale collabito può assumere un aspetto a bulbo, solitamente suggestivo degli esiti di un’invaginazione. Anche in questo tipo di atresia la lunghezza intestinale è praticamente normale.
TIPO III
- III a (16%): è presente una soluzione di continuità tra i due segmenti intestinali ed un difetto del mesentere di dimensioni variabili. Il tratto intestinale prossimale, dilatato, a fondo cieco è spesso aperistaltico e frequentemente può andare incontro a torsione o divenire iperdisteso, con successiva necrosi e perforazione quale evento secondario. In questo tipo di atresia la lunghezza totale dell’intestino è variabile ma solitamente inferiore alla normalità.
- III b (19%) nota anche come atresia apple-peel o Christmas-tree: è caratterizzata da un’atresia digiunale alta, vicino al legamento di Treitz, con lunghezza ridotta dell’intestino ed un ampio difetto mesenterico in cui è coinvolta la radice dell’arteria mesenterica superiore, che resta pervia distalmente, alimentata in senso retrogrado dalla circolazione nella arteria ileocolica. Il tratto intestinale distale collabito si presenta libero in addome e presenta uno sviluppo elicoidale su di un breve e precario supporto vascolare derivato dal ramo ileocolico alimentato dall’arcata colica destra o dalla mesenterica inferiore. Questa disposizione dell’intestino distale può ricordare appunto la buccia di una mela appena sbucciata o l’abete natalizio. Questo tipo di atresia comporta sempre una lunghezza totale dell’intestino inferiore alla norma.
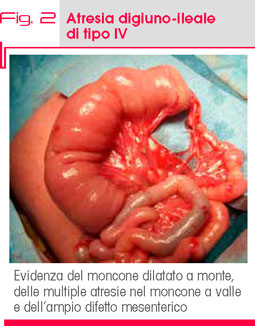 TIPO IV (6-20%): coesistenza di multiple atresie intestinali, soprattutto di tipo I e tipo II (Fig. 2).
TIPO IV (6-20%): coesistenza di multiple atresie intestinali, soprattutto di tipo I e tipo II (Fig. 2).
L’atresia di tipo IV si associa molto spesso a prematurità e determina sempre una ridotta estensione del tratto intestinale. I neonati affetti da questa forma presentano lunghi segmenti di piccolo e grosso intestino completamente occlusi senza un lume riconoscibile. Un incidente vascolare prenatale responsabile di un così esteso coinvolgimento del tratto gastrointestinale ha fatto supporre il ruolo determinante di un fattore genetico a trasmissione autosomica recessiva anche se la maggior parte dei casi con atresia intestinale multipla sono sporadici e non presentano altri difetti gastrointestinali associati.
E’ descritta l’associazione tra atresia di tipo IV e immunodeficienze severe le cui conseguenze compromettono la sopravvivenza della maggior parte dei pazienti oltre i due anni di età (10).
Le atresie del piccolo intestino sono equamente distribuite fra digiuno e ileo: statisticamente la sede più coinvolta è l’ileo terminale (36%), seguita dal digiuno prossimale (31%), dal digiuno distale (20%) e dall’ileo prossimale (13%).
L’atresia digiuno-ileale è un’entità per lo più isolata (>90%), sebbene il 6-20% dei casi possa presentarsi con atresie multiple per lo più coinvolgenti il digiuno prossimale. Rarissima l’associazione con quadri sindromici e, se si escludono le malformazioni dell’apparato digerente (malrotazioni intestinali, volvolo intestinale, invaginazione, malformazioni ano-rettali) e della parete addominale (onfalocele, gastroschisi), eccezionali sono le anomalie associate di altri organi e/o apparati (anomalie vertebrali, cardiopatie congenite, difetti del tubo neurale), sia poiché l’atresia si sviluppa tardivamente nel corso della vita fetale sia in quanto il danno vascolare è circoscritto.
Terapia
La terapia dell’atresia digiuno-ileale è chirurgica. Mentre le forme di tipo I e II richiedono solo il ripristino della continuità intestinale, è evidente che le atresie di tipo III e di tipo IV sono più complesse per il coinvolgimento più esteso del canale alimentare, e pongono quindi i maggiori problemi per gli esiti riguardo alla lunghezza dell’intestino residuo.
La tecnica operativa utilizzata per ciascun paziente affetto da atresia digiuno-ileale va individualizzata a seconda della severità del difetto da correggere e della eventuale coesistenza di complicazioni o altre patologie associate quali malrotazione, volvolo, ileo da meconio o peritonite meconiale, che possono modificare l’indicazione terapeutica. Anche la presenza di un difetto della parete addominale (gastroschisi, onfalocele) e le contestuali caratteristiche dell’intestino, nonché le condizioni generali e la stabilità emodinamica del paziente influenzano la scelta chirurgica.
Storicamente è noto che l’utilizzo del moncone atresico prossimale dilatato per confezionare una anastomosi termino-terminale fra monconi incongruenti spesso esita in un’ostruzione funzionale, mentre un’anastomosi intestinale latero-laterale, ampiamente utilizzata in età adulta per ovviare al problema, esita sia in un’ostruzione funzionale che in una sindrome dell’ansa cieca.
L’ampia discrepanza di calibro tra il segmento atresico prossimale e quello distale, la dismotilità e la distrofia del tratto dilatato spiegano il motivo per cui spesso un’anastomosi primaria non è funzionale ed i pazienti manifestano intolleranza alimentare, ostruzione intestinale funzionale e aumento della proliferazione batterica con conseguente scarso adattamento dell’intestino e segni di malassorbimento (18). Per rimediare alla discrepanza di lume e ottimizzare la funzionalità intestinale, sono state proposte varie tecniche chirurgiche:
- resezione in toto del tratto prossimale più dilatato;
- enteroplastica longitudinale (tapering) mediante sezione della parete intestinale a tutto spessore o del solo strato siero muscolare sul versante antimesenterico;
- plicatura intestinale con introflessione del tratto dilatato all’interno del lume intestinale.
Mentre la plicatura è gravata da un’alta percentuale di insuccesso, sia la resezione intestinale che l’enteroplastica causano una perdita della superficie mucosa assorbente, già congenitamente ridotta nei tipi di atresia complessa (tipo III e IV). In questi casi dunque la terapia stessa può contribuire a determinare una ulteriore grave complicanza dell’atresia digiuno-ileale, ossia la sindrome da malassorbimento per intestino corto, i cui effetti e morbilità possono essere estremamente gravi.
Sindrome dell’intestino corto
La sindrome dell’intestino corto (Short Bowel Syndrome - SBS), sinonimo di insufficienza intestinale, è caratterizzata da un malassorbimento intestinale secondario alla perdita massiva di superficie assorbente di piccolo intestino a causa di patologie congenite o acquisite. Manifestazioni cliniche della SBS sono la disidratazione secondaria a diarrea, la mancata crescita e le stigmate della malnutrizione proteico-calorica fino alle conseguenze più estreme (11).
 Sebbene modelli animali con SBS siano definiti da una mancanza di piccolo intestino dell’80% o oltre (32), nell’uomo non si è arrivati a porre una misura limite, e per convenzione la SBS viene correlata direttamente alla dipendenza dalla nutrizione parenterale (NP), unica possibilità di sopravvivenza per questi soggetti. Come riportato da Goulet e collaboratori, l’atresia intestinale è tra i principali fattori predittivi associati a più alta percentuale di dipendenza da NP a lungo termine (Tab. 1).
Sebbene modelli animali con SBS siano definiti da una mancanza di piccolo intestino dell’80% o oltre (32), nell’uomo non si è arrivati a porre una misura limite, e per convenzione la SBS viene correlata direttamente alla dipendenza dalla nutrizione parenterale (NP), unica possibilità di sopravvivenza per questi soggetti. Come riportato da Goulet e collaboratori, l’atresia intestinale è tra i principali fattori predittivi associati a più alta percentuale di dipendenza da NP a lungo termine (Tab. 1).
Normalmente la lunghezza del piccolo intestino è riportata essere di 200-250 cm nei neonati a termine mentre nei prematuri varia da 160 a 240 cm; anche questa variabilità contribuisce a rendere sfumati i limiti della patologia da SBS. Recenti revisioni sull’argomento sostengono che pazienti con meno di 35 cm di piccolo intestino sono ad alto rischio di dipendenza dalla NP a lungo termine, mentre d’altra parte sono stati descritti casi di bambini con circa 10 cm di intestino che hanno potuto essere svezzati rapidamente dalla NP (12). Esistono infatti diversi fattori, oltre alla superficie assorbente, che possono influenzare la dipendenza o meno dalla NP, come una scarsa motilità intestinale, un malassorbimento nell’intestino residuo o anche un transito troppo rapido.
Sotto questo aspetto, la prematurità invece sembrerebbe correlare con una prognosi migliore per i pazienti con SBS da atresia del tenue, per il loro maggiore potenziale di crescita che consentirebbe all’intestino di svilupparsi in lunghezza e quindi di fornire un buon adattamento e un’adeguata funzione di assorbimento a distanza di tempo.
Anche la presenza di una valvola ileo-ciecale, per il suo contributo al mantenimento di una fisiologica ecologia intestinale, è ritenuta un fattore prognostico favorevole in pazienti con SBS in termini di possibilità di svezzamento dalla NP.
La NP a lungo-termine, indispensabile procedura salva-vita per il paziente con SBS, non sempre si dimostra una risorsa percorribile per tempi indefiniti, sia per l’insorgenza di gravissime complicanze, come la colestasi (Parenteral Nutrition Associated Cholestasis - PNAC), sia per l’esaurimento degli accessi venosi.
In particolare la colestasi e la malattia epatica correlate alla NP (PNALD) rappresentano le peggiori complicanze e si associano ad alta mortalità (13). In particolare, la malattia epatica, nota anche come IFALD (Intestinal Failure Associated Liver Disease), interessa il 40-60% dei pazienti e sino al 16.6% dei casi può progredire verso un’insufficienza epatica irreversibile (14). L’eziologia della PNAC e della PNALD è multifattoriale; fattori di rischio noti includono la prematurità, il basso peso alla nascita, la mancanza di nutrizione enterale, la sepsi, procedure chirurgiche multiple, la durata della NP e, correlata a quest’ultima, la composizione delle soluzioni lipidiche utilizzate. In particolare diversi lavori su modelli animali e, più recentemente, alcuni lavori clinici hanno riportato quale prevenzione e reversibilità della IFALD la sostituzione delle tradizionali emulsioni lipidiche a base di soia contenenti acidi grassi omega-6 (Ω-6), con emulsioni lipidiche a base di olio di pesce ricchi in acidi grassi Ω-3. Queste ultime infatti riducono i processi infiammatori responsabili della progressiva malattia epatica che gradualmente evolve verso l’insufficienza d’organo (15).
Anche se alcune di queste complicanze attualmente sono meglio trattate e prevenute, l’evoluzione clinica dei pazienti con gravi esiti di atresia digiuno-ileale complessa risulta strettamente dipendente dalle limitate possibilità fisiche di mantenere un catetere venoso centrale e una NP a lungo termine.
Nella gestione multidisciplinare dell’insufficienza intestinale la chirurgia svolge un ruolo fondamentale, e data la severità del percorso di cura che può arrivare al trapianto multiorgano (16), ogni scelta deve essere considerata con lungimiranza, fin dall’iniziale correzione della patologia malformativa in epoca neonatale.
Possibilità di trattamento della SBS
Nel trattamento dell’insufficienza intestinale i risultati ancora insoddisfacenti del trapianto di intestino giustificano i vari sforzi terapeutici alternativi che, tuttavia, vengono presi in considerazione solo quando la SBS è conclamata, ben oltre il periodo neonatale.
Fra questi i principali attualmente perseguiti cercano di sfruttare la dilatazione compensatoria del tratto intestinale residuo per rimodellare la superficie disponibile in un canale alimentare di minor diametro e maggiore lunghezza allo scopo di aumentare il tempo di contatto delle sostanze alimentari con la mucosa intestinale assorbente.
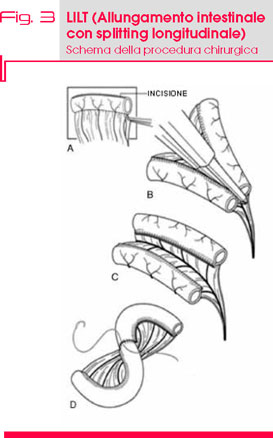 Nel 1980 è stato descritto da Bianchi l’allungamento intestinale longitudinale (LILT, longitudinal intestinal lengthening and tailoring) (17) (Fig. 3), che prevede il raddoppio della lunghezza di un segmento intestinale a scapito del diametro, separando la vascolarizzazione più terminale del rispettivo mesentere. Questa tecnica non aumenta la superficie mucosa assorbente ma migliora la funzionalità del piccolo intestino residuo.
Nel 1980 è stato descritto da Bianchi l’allungamento intestinale longitudinale (LILT, longitudinal intestinal lengthening and tailoring) (17) (Fig. 3), che prevede il raddoppio della lunghezza di un segmento intestinale a scapito del diametro, separando la vascolarizzazione più terminale del rispettivo mesentere. Questa tecnica non aumenta la superficie mucosa assorbente ma migliora la funzionalità del piccolo intestino residuo.
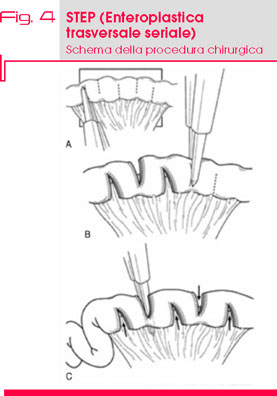 Nel 2003, è stata proposta una tecnica alternativa di allungamento e rimodellamento intestinale, chiamata STEP (Serial Transverse Enteroplasty Procedure) (18) (Fig. 4) e descritta per la prima volta su un modello bovino e successivamente sul bambino.
Nel 2003, è stata proposta una tecnica alternativa di allungamento e rimodellamento intestinale, chiamata STEP (Serial Transverse Enteroplasty Procedure) (18) (Fig. 4) e descritta per la prima volta su un modello bovino e successivamente sul bambino.
A differenza della LILT questa procedura non richiede una dilatazione intestinale uniforme, preserva l’apporto vascolare naturale dell’intestino e non richiede la necessità di eseguire enterotomie, evitando quindi il rischio di contaminazione batterica del cavo peritoneale. Per questi motivi l’applicazione della STEP è andata aumentando in ambito chirurgico pediatrico come trattamento rescue in caso di SBS con dilatazione ileale.
L’applicazione di queste nuove tecniche chirurgiche ha permesso un aumento della tolleranza alla nutrizione enterale ed la conseguente riduzione della necessità della NP, contribuendo in molti casi alla risoluzione della insufficienza intestinale da SBS.
In tempi molto recenti la tecnica STEP è stata applicata in casi aneddotici direttamente anche in epoca neonatale, in sostituzione delle enteroplastiche (19).
Caso clinico
Un neonato maschio di 37 settimane di età gestazionale, nato da parto eutocico, con peso alla nascita di 2710 gr, giungeva alla nostra attenzione per vomito biliare comparso in prima giornata di vita. Dall’anamnesi pre-natale emergeva un riscontro ecografico di polidramnios e di distensione di poche anse intestinali, reperti suggestivi per atresia intestinale digiuno-ileale. L’Rx addome diretto dimostrava una marcata distensione gassosa dello stomaco e di alcune anse del piccolo intestino senza presenza di aria nei quadranti addominali inferiori. Alla luce del quadro clinico-strumentale che supportava il sospetto diagnostico prenatale, si rendeva necessario procedere a laparotomia esplorativa.
Come primo reperto intraoperatorio si riscontrava la presenza di un’ansa digiunale notevolmente dilatata ed ipertrofica con un piccolo moncone efferente non comunicante a fondo cieco. Successivamente a tale riscontro, si constatava, pochi centimetri a valle del digiuno atresico, un segmento intestinale ostruito da membrana e dopo circa cinque centimetri una seconda membrana cui seguiva l’ultimo tratto digiunale quasi completamente atresico con mesentere ipotrofico, mentre l’ileo si presentava interamente pervio. Veniva inoltre individuata una malrotazione intestinale. Si poneva pertanto diagnosi di atresia intestinale digiuno-ileale multipla complessa, associata a malrotazione intestinale.
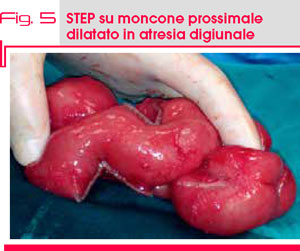 In considerazione della notevole dilatazione del tratto digiunale atresico prossimale e della compromissione anche del tratto a valle della prima atresia, si decideva di eseguire due procedure di STEP, allo scopo di rendere i monconi da anastomizzare più congruenti senza sacrificare una parte del moncone digiunale dilatato (Fig. 5).
In considerazione della notevole dilatazione del tratto digiunale atresico prossimale e della compromissione anche del tratto a valle della prima atresia, si decideva di eseguire due procedure di STEP, allo scopo di rendere i monconi da anastomizzare più congruenti senza sacrificare una parte del moncone digiunale dilatato (Fig. 5).
Non è stato possibile eseguire ulteriori procedure di STEP sul tratto più prossimale dell’ansa dilatata perché costituito dal duodeno, completamente endoperitoneale per la malrotazione e quindi con il rischio di compromettere l’integrità del coledoco. Si procedeva, inoltre, a sezione delle due atresie di tipo I (membrane digiunali) attraverso piccole enterotomie e a confezionamento di due anastomosi rispettivamente digiuno-digiunale e digiuno-ileale. Infine si posizionava la matassa intestinale a mesenterium commune.
Nel periodo post-operatorio persistenti difficoltà del transito intestinale hanno richiesto un secondo intervento per lisi di aderenze intraddominali e revisione delle pregresse membrane endoluminali, mentre l’ansa trattata con STEP non presentava nessun problema di transito. Tra il primo ed il secondo intervento chirurgico il paziente veniva supportato con NP totale che gli permetteva di mantenere un equilibrio idro-elettrolitico ed uno stato nutrizionale adeguato pur con una iniziale alterazione degli indici di colestasi epatica (bilirubina diretta 4.5 mg/dl e gammaGT 392 U/I).
In seguito alla revisione chirurgica, la rapida ricanalizzazione intestinale ha consentito l’inizio ed il progressivo aumento della nutrizione enterale e la graduale riduzione della NP con normalizzazione della colestasi nell’arco di due mesi (bilirubina diretta 0.19 mg/dl e gammaGT 113 U/I).
Il decorso clinico successivo ha confermato una buona tolleranza alla nutrizione per via orale con latte materno misto a latte formulato ed una sostanziale regolarità delle evacuazioni per quantità e frequenza.
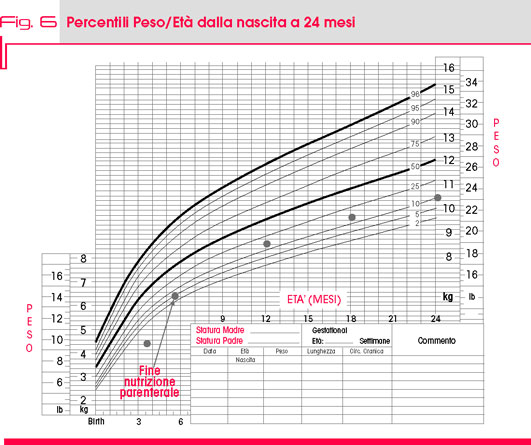 Il paziente veniva quindi dimesso all’età di tre mesi e mezzo con un peso di 4490 gr, con integrazione di un modico apporto di nutrizione parenterale domiciliare sospesa definitivamente dopo due mesi dalla dimissione alla luce di una ripresa sicura della crescita ponderale (Fig. 6).
Il paziente veniva quindi dimesso all’età di tre mesi e mezzo con un peso di 4490 gr, con integrazione di un modico apporto di nutrizione parenterale domiciliare sospesa definitivamente dopo due mesi dalla dimissione alla luce di una ripresa sicura della crescita ponderale (Fig. 6).
In seguito, il paziente veniva sottoposto a controlli clinici ambulatoriali, con frequenza mensile nel primo anno dalla dimissione, poi ogni tre mesi nel secondo anno, ed in seguito ogni sei mesi negli anni successivi.
Dalla dimissione non si sono mai verificati episodi di occlusione intestinale.
Il paziente ha continuato a presentare buona tolleranza alla nutrizione orale con normale introduzione dello svezzamento e progressivo ampliamento della dieta alimentare, mantenendo sempre un alvo con evacuazioni nella norma per frequenza e consistenza.
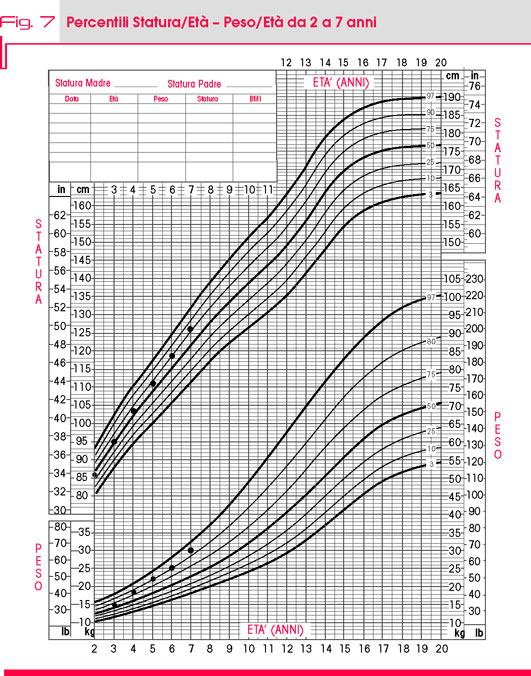 La curva di crescita staturo-ponderale, caratterizzata nei primi due anni di vita da percentili sempre compresi tra il 10° ed il 50° (Fig. 6), è arrivata in seguito ad oscillare tra il 50° e il 75° percentile, con la crescita staturale saldamente intorno al 50° percentile. All’ultimo controllo clinico, a sette anni dall’intervento, peso e altezza sono stati rilevati rispettivamente tra il 90° ed il 97° percentile, e al 75° percentile (Fig. 7).
La curva di crescita staturo-ponderale, caratterizzata nei primi due anni di vita da percentili sempre compresi tra il 10° ed il 50° (Fig. 6), è arrivata in seguito ad oscillare tra il 50° e il 75° percentile, con la crescita staturale saldamente intorno al 50° percentile. All’ultimo controllo clinico, a sette anni dall’intervento, peso e altezza sono stati rilevati rispettivamente tra il 90° ed il 97° percentile, e al 75° percentile (Fig. 7).
Considerazioni conclusive
L’atresia congenita digiuno-ileale è una patologia rara che si manifesta fin dall’epoca perinatale con un ampio spettro di quadri clinici, in cui le forme più complesse (tipo IIIa, IIIb e IV) possono determinare una SBS e porre seri problemi di trattamento a lungo termine. La terapia è essenzialmente chirurgica e deve tenere conto di tutte le possibili implicazioni future, fin dal primo atto della correzione in epoca neonatale.
Il caso clinico riportato esemplifica questa complessità per il quadro patologico caratterizzato sia dalla coesistenza di atresie multiple, che implica un deficit della lunghezza intestinale complessiva, che dalla mancata rotazione intestinale.
In questo caso il trattamento tradizionale, concentrandosi sul tentativo di confezionare un’anastomosi congruente, avrebbe comportato una importante ulteriore riduzione della capacità di assorbimento ed il rischio quindi di incorrere in una SBS.
Con lo scopo di preservare tutto l’intestino disponibile al fine di prevenire la dipendenza dalla NP, è stato deciso di applicare direttamente in epoca neonatale la tecnica STEP di allungamento e rimodellamento intestinale, per quanto consentito dalla malrotazione.
Questa procedura, ancora eccezionalmente utilizzata nel neonato, anche per limiti imposti dalla tecnologia, ha permesso di rimodellare l’ansa dilatata ed ipertrofica e contemporaneamente di risolvere la discrepanza di calibro tra i due monconi atresici, con il vantaggio di realizzare un’anastomosi intestinale termino-terminale, di mantenere la funzionalità motoria del tratto prossimale e, eliminando il ristagno, di ridurre la proliferazione batterica. Tutto questo ha dimostrato di riflettersi positivamente sull’assorbimento intestinale e quindi sul contenimento della necessità di supporto NP di lunga durata.
Qualora esperienze analoghe venissero confermate da casistiche più vaste, si potrebbe segnare un passo importante nel trattamento delle atresie digiuno-ileali più severe e nella prevenzione di una delle principali cause di SBS.
La nostra esperienza, come già riportato da alcune segnalazioni in letteratura, indica come l’applicazione senza preconcetti di tecniche chirurgiche innovative possa contribuire a importanti progressi nel trattamento di malformazioni congenite complesse e nella prevenzione delle loro conseguenze ed esiti a più lungo termine.
Bibliografia
- Mishalany, H. G., Najjar, F. B. Familial jejunal atresia: three cases in one family. J. Pediat. 73: 753-755, 1968.
- Shorter NA, Georges A, Perenyib A, et al. A proposed classification system for familial intestinal atresia and its relevance to the understanding of the etiology of jejunoileal atresia. Journal of Pediatric Surgery (2006) 41, 1822–1825.
- Seashore JH, Collins FS, Markowitz RI, Seashore MR. Familial apple peel jejunal atresia: surgical, genetic, and radiographic aspects. Pediatrics. 1987;80:540-4.
- Samuels ME, Majewski J, Alirezaie N, et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J Med Genet. 2013;50(5):324-9.
- Bigorgne AE, Farin HF, Lemoine R, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest. 2014;124(1):328-37.
- Filges I, Bruder E, Brandal K, et al. Strømme Syndrome Is a Ciliary Disorder Caused by Mutations in CENPF. Hum Mutat. 2016;37(4):359-63.
- Stollman TH, de Blaauw I, Wijnen MH, et al. Decreased mortality but increased morbidity in neonates with jejunoileal atresia; a study of 114 cases over a 34-year period. J Pediatr Surg 44:217-221, 2009
- Roberts HE, Cragan JD, Cono J, et al. A Increased frequency of cystic fibrosis among infants with jejunoileal atresia. Am J Med Genet. 1998 6;78(5):446-9
- Grosfeld JL: Jejunoileal atresia and stenosis. In: “Pediatric Surgery”, Year Book Medical Publishers, inc. Chicago, London 1986
- Notarangelo LD. Multiple intestinal atresia with combined immune deficiency. Curr Opinion Pediatr, 2014; 26 (6): 690-6
- Vanderhoof JA et al. Short bowel syndrome in children and adults. Gastroenterol 1997; 113: 1767-78
- Buchman AL. Etiology and initial management of short bowel syndrome. Gastroenterology 2006; 130 (Suppl): S5-15
- Christensen RD et al: Identifying patients, on the first day of life, at high-risk of developing parenteral nutrition-associated liver disease. J Perinatol. 27:284-290 2007
- Yang CF et al.: Persistent alanine aminotransferase elevations in children with parenteral nutrition-associated liver disease. J Pediatr Surg. 44:1084-1087 2009
- Shin JI et al. Could lipid infusion be a risk for parenteral nutrition-associated cholestasis in low birth weight neonates? Eur J Pediatr 2008; 167: 197-202
- Biren P Modi et al. First report of the International Serial Transverse Enteroplasty Data Registry: indications, efficacy, and complications. J Am Coll Surg, 2007. 365-371/
- Bianchi A. Intestinal loop lengthening- a technique for increasing small intestinal length. J Pediatr Surg, 1980; 15: 145-151
- Kim H et al. Serial transverse enteroplasty: a novel bowel lengthening procedure. J Pediatr Surg, 2003; 38: 425-429
- Wales PW et al. Serial transverse enteroplasty as primary therapy for neonates with proximal jejunal atresia. J Pediatr Surg 2005; 40: E31-4