




la Newsletter

Silvia Pontesilli, Cristina Baldoli
Unità di Neuroradiologia IRCCS San Raffaele - Milano
MPS tipo I: ruolo della risonanza magnetica cerebrale e spinale...
La Mucopolisaccaridosi di tipo I, o sindrome di Hurler, è una...
 La Mucopolisaccaridosi (MPS) di tipo I o Sindrome di Hurler, è una malattia genetica da accumulo lisosomiale di glicosaminoglicani a livello di organi, sistema nervoso centrale (SNC) e scheletro (1). Il coinvolgimento cerebrale porta a ritardo/regressione cognitiva, l’anomalo sviluppo osseo coinvolge anche il rachide con deformità e compressione midollare (2). La Risonanza Magnetica (RM) consente di valutare in modo non invasivo tali quadri (3,4,5,6).
La Mucopolisaccaridosi (MPS) di tipo I o Sindrome di Hurler, è una malattia genetica da accumulo lisosomiale di glicosaminoglicani a livello di organi, sistema nervoso centrale (SNC) e scheletro (1). Il coinvolgimento cerebrale porta a ritardo/regressione cognitiva, l’anomalo sviluppo osseo coinvolge anche il rachide con deformità e compressione midollare (2). La Risonanza Magnetica (RM) consente di valutare in modo non invasivo tali quadri (3,4,5,6).
A livello cerebrale la RM mostra peculiare conformazione cranica, macrocrania con ispessimento della diploe, platibasia con stenosi del forame magno, dismorfismo del cavo sellare. Inoltre è possibile valutare il trofismo cerebrale, parametro correlato allo sviluppo cognitivo, misurando le dimensioni di sistema ventricolare e spazi subaracnoidei, per lo più ampliati.
Peculiare è il riscontro di spazi perivascolari (SPV) ampliati ubiquitari nella sostanza bianca, frequente il coinvolgimento del corpo calloso. La sostanza bianca può presentare alterazioni focali, spesso in corrispondenza degli SPV (Fig. 1).
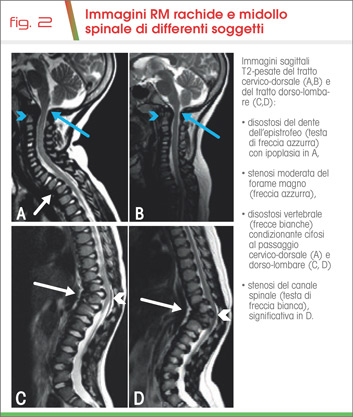
A livello rachideo con la RM è possibile valutare l’anomalo sviluppo osseo vertebrale. I somi sono deformati con conseguente tipica angolatura cifotica dorso-lombare e possibile cervico-dorsale; tale deformazione su base disostosica può condizionare una stenosi del canale spinale. Caratteristico è l’anomalo sviluppo del dente dell’epistrofeo che risulta ipoplasico, con ipertrofia legamentosa e instabilità, stenosi del forame magno e possibile mielopatia secondaria (Fig. 2).
 Uno specifico score (Tab. 1a, 1b) raggruppa i parametri RM e ci consente di valutare in modo semiquantitativo il coinvolgimento patologico all’esordio e monitorarlo nel tempo, anche in relazione ai trattamenti eseguiti (7). Oltre al gold standard rappresentato dal trapianto di midollo osseo (8,9), la terapia genica in via sperimentale si sta rivelando promettente nel rallentare l’andamento della malattia e consentire una migliore qualità di vita a questi piccoli pazienti (10).
Uno specifico score (Tab. 1a, 1b) raggruppa i parametri RM e ci consente di valutare in modo semiquantitativo il coinvolgimento patologico all’esordio e monitorarlo nel tempo, anche in relazione ai trattamenti eseguiti (7). Oltre al gold standard rappresentato dal trapianto di midollo osseo (8,9), la terapia genica in via sperimentale si sta rivelando promettente nel rallentare l’andamento della malattia e consentire una migliore qualità di vita a questi piccoli pazienti (10).
Lo score di valutazione raggruppa le principali caratteristiche della malattia riconoscibili con le immagini di RM. Ad ogni parametro di coinvolgimento del SNC (tab. 1a e 1b) viene attribuito un punteggio crescente di interessamento, secondo valutazioni il più possibile quantitative e ripetibili, in questo modo la somma dei punteggi definisce la gravità di espressione della malattia a livello del SNC. Lo score è strutturato in modo da poter analizzare separatamente il risultato di coinvolgimento dei due compartimenti del SNC, encefalico e spinale, questo ci consente di correlare i risultati con i diversi aspetti della malattia, lo sviluppo cognitivo per l’encefalo e lo sviluppo osseo per il rachide e midollo spinale (7). Inoltre permette una migliore valutazione della risposta alla terapia nel follow-up.


Bibliografia
- Clarke LA. Mucopolysaccharidosis Type I. 2002 Oct 31 [updated 2021 Feb 25]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. PMID: 20301341.
- Bigger BW, et al. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol Genet Metab. 2018;125(4):322-331.
- Reichert R, et al. Neuroimaging Findings in Patients with Mucopolysaccharidosis: What You Really Need to Know. Radiographics. 2016;36(5):1448-62.
- Schmidt M, et al. Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J Rare Dis. 2016;11(1):93.
- Nicolas-Jilwan M, AlSayed M. Mucopolysaccharidoses: overview of neuroimaging manifestations. Pediatr Radiol. 2018;48(10):1503-1520.
- Tandon V, Williamson JB, Cowie RA, Wraith JE. Spinal problems in mucopolysaccharidosis I (Hurler syndrome). J Bone Joint Surg Br. 1996;78(6):938-44.
- Pontesilli S, Baldoli C, ... , Parini R. Evidence of Treatment Benefits in Patients with Mucopolysaccharidosis Type I-Hurler in Long-term Follow-up Using a New Magnetic Resonance Imaging Scoring System. J Pediatr. 2022;240:297-301.e5.
- Wang RY, et al. Treatment reduces or stabilizes brain imaging abnormalities in patients with MPS I and II. Mol Genet Metab. 2009;98(4):406-11.
- Manara R, et al. Brain and spine MRI features of Hunter disease: frequency, natural evolution and response to therapy. J Inherit Metab Dis. 2011;34(3):763-80.
- Gentner B, et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N Engl J Med. 2021;385(21):1929-1940.