




la Newsletter

Simona Bianchi, Fiorina Giona
Ematologia, Dipartimento di Medicina Traslazionale e di Precisione, Sapienza Università di Roma
Malattia di Castleman: un enigma diagnostico e terapeutico | La...
La malattia di Castleman comprende un gruppo eterogeneo di...
Nel 1991 giungeva alla nostra osservazione un ragazzo di 17 anni, per dolori addominali ricorrenti, associati a stranguria ed ematuria. Le indagini di laboratorio escludevano patologie ematologiche o infettive. La TC mostrava adenopatie multiple mediastiniche, un linfonodo sovraclaveare destro e marcata splenomegalia. La biopsia linfonodale permetteva di fare diagnosi di malattia di Castleman angiofollicolare, variante plasmacellulare.
Veniva trattato con a-Interferon (IFN-a), sospeso nel 1996 per scarsa compliance, ma con miglioramento del quadro radiologico. Nel gennaio 2000, per l’incremento della splenomegalia veniva sottoposto a splenectomia, il cui esame istologico deponeva per un quadro tipico di malattia di Castleman (MC). Nel maggio 2008, per comparsa di linfoadenopatia ascellare destra, veniva effettuata una nuova biopsia che confermava la diagnosi di malattia di Castleman, variante ialino-vascolare. Nell’aprile 2010, per una piastrinopenia isolata (PLT 47.000/mmc) non secondaria a malattia linfoproliferativa, accompagnata da sintomatologia emorragica, veniva iniziata terapia con basse dosi di prednisone con normalizzazione della conta piastrinica. Nel novembre 2020, all’età di 46 anni, per la comparsa di diarrea non causata da patologie intestinali, l’aumento delle linfoadenopatie e degli indici di flogosi (VES 34, PCR 13500 mcg/ml e IL-6 31,28 pg/ml) iniziava terapia specifica con anti-Interleuchina-6 (IL-6) (siltuximab) alla dose standard, con scomparsa della sintomatologia addominale, normalizzazione della PCR dopo due dosi, e progressiva riduzione dei livelli di IL-6.
Patogenesi e manifestazioni cliniche
La malattia di Castleman comprende un gruppo eterogeneo di disordini, caratterizzati dalla presenza di adenomegalia accompagnata o meno da sintomi sistemici, e da un andamento clinico variabile. La MC può colpire tutte le età, con manifestazioni ed andamento clinico variabili. Il meccanismo patogenetico comune sembrerebbe legato ad una attivazione di uno stato infiammatorio per aumentata produzione e rilascio in circolo di citochine pro-infiammatorie, tra cui l’IL-6 sembra avere un ruolo centrale (1). In base al numero delle stazioni linfonodali coinvolte, sono state identificate due forme di MC (2):
- Malattia di Castleman Unicentrica(MCU), limitata a una singola stazione linfonodale, non associata ad infezioni da HIV e/o HHV8;
- Malattia di Castleman Multicentrica(MCM), caratterizzata da linfoadenopatie multiple. Comprende: la forma associata a HHV8 (MCM HHV8+), la forma HHV8 negativa (MCM HHV8-), che, a sua volta, comprende la forma classica, idiopatica (iMCM), quella idiopatica non altrimenti specificata (iMCM-NOS), quella associata ad una disfunzione d’organo, la iMCM-TAFRO (trombocitopenia, anasarca, febbre, disfunzione renale e organomegalia); e la forma caratterizzata da polineuropatia, organomegalia, endocrinopatia, componente monoclonale, alterazioni cutanee (skin), definita MCM-POEMS.
La presentazione clinica è variabile, da casi completamente asintomatici a quelli con sintomi sistemici e/o coinvolgimento d’organo. Nella iMCM, oltre alle linfoadenopatie multiple, sono presenti epato e/o splenomegalia, febbre, citopenia e alterazione della funzionalità epatorenale (1). Nelle forme iMCM-TAFRO e MCM-POEMS, che è una sindrome paraneoplastica, il quadro clinico è caratterizzato da una compromissione multiorgano.
Diagnosi
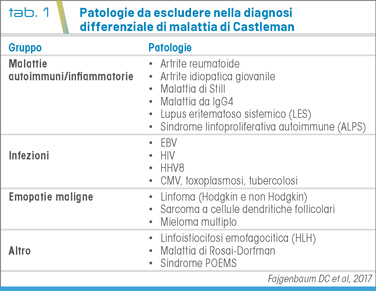 La MC entra in diagnosi differenziale con malattie autoimmuni/disreattive, infettive ed emopatie maligne (Tab. 1).
La MC entra in diagnosi differenziale con malattie autoimmuni/disreattive, infettive ed emopatie maligne (Tab. 1).
L’esame istologico e immunoistochimico linfonodale con la ricerca di HHV8 e HIV è indispensabile per la diagnosi di certezza (1). La caratterizzazione istologica permette di identificare quattro istotipi: variante ialino-vascolare, variante ipervascolare, variante plasmacellulare, variante mista (3). La valutazione di alcuni criteri, clinici e di laboratorio (Tab. 2) è indispensabile per la definizione della forma iMCM, la cui diagnosi richiede la presenza di entrambi i criteri clinici maggiori e di almeno 2/11 criteri minori (4).
Trattamento
Il trattamento standard della MCU è l’escissione chirurgica completa; laddove non sia possibile, si ricorre a trattamenti locali o sistemici, in caso di uno stato infiammatorio acuto (5).
 Per la iMCM HHV8-, il farmaco d’elezione in prima linea è siltuximab (11 mg/kg/dose ogni 3 settimane) (6); nei casi molto gravi, con sintomatologia sistemica, le dosi di siltuximab sono settimanali ed associate ad alte dosi di steroidi (2,8). Nella MCM HHV8+/HIV-, la terapia di scelta è rituximab, associato o meno a chemioterapia; in alcuni casi si è dimostrata efficace anche la terapia antivirale con valganciclovir, in associazione alla chemio e/o immunoterapia (7,3,8). I pazienti con iMCM HHV8-, resistenti a siltuximab, dopo una nuova biopsia per conferma diagnostica, hanno diverse opzioni terapeutiche: corticosteroidi, immunosoppressori, anticorpi monoclonali, interferone e schemi chemioterapici lymphoma-like oppure myeloma-like (9).
Per la iMCM HHV8-, il farmaco d’elezione in prima linea è siltuximab (11 mg/kg/dose ogni 3 settimane) (6); nei casi molto gravi, con sintomatologia sistemica, le dosi di siltuximab sono settimanali ed associate ad alte dosi di steroidi (2,8). Nella MCM HHV8+/HIV-, la terapia di scelta è rituximab, associato o meno a chemioterapia; in alcuni casi si è dimostrata efficace anche la terapia antivirale con valganciclovir, in associazione alla chemio e/o immunoterapia (7,3,8). I pazienti con iMCM HHV8-, resistenti a siltuximab, dopo una nuova biopsia per conferma diagnostica, hanno diverse opzioni terapeutiche: corticosteroidi, immunosoppressori, anticorpi monoclonali, interferone e schemi chemioterapici lymphoma-like oppure myeloma-like (9).
Conclusioni
Essendo una malattia molto rara ed eterogenea, il modello di ricerca di tipo tradizionale è stato il principale fattore limitante lo sviluppo delle conoscenze sulla patologia, fino a un decennio fa. La creazione, nel 2012, del Castleman Disease Collaborative Network (CDCN) (www.cdcn.org) ha dato un notevole impulso allo sviluppo della ricerca clinica e di laboratorio, soprattutto nella forma iMCM HHV8-, che ha un andamento particolarmente aggressivo (10). Lo sforzo condiviso ha portato, nel 2016, alla creazione di un Registro della Malattia di Castleman (ACCELERATE), in cui sono coinvolti direttamente i pazienti (10). Successivamente, è stata istituita una biobanca, la Castleman Disease Biobank (Castlebank), in cui vengono collezionati campioni di sangue e/o tessuto, in modo da poter sviluppare una ricerca clinica e di laboratorio che potrà portare nel prossimo futuro a importanti miglioramenti, soprattutto nelle forme più rare e complesse.
Bibliografia
- Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control. 2014; 21: 266-278.
- Phulware RH, Ramteke P, Yadav R, et al. Cytology of Castleman’s disease (hyaline-vascular type) masquerading as Hodgkin’s lymphoma Am J Blood Res. 2022;12(6):196-200.
- Yoshizaki K, Murayama S, Ito H, Koga T. The Role of Interleukin-6 in Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):23-36.
- He L, Chen Y, Tan X, et al. 18F-FDG PET/CT and contrast-enhanced CT in the diagnosis of Castleman disease. Jpn J Radiol. 2023;41(1):98-107.
- van rhee F, Greenway A, Stone K. Treatment of Idiopathic Castleman Disease. Hematol Oncol Clin N Am, 2018;32:89-106.
- Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidenced-based consensus diagnostic criteria for HHV-8 – negative/idiopathic multicenter Castleman disease. Blood. 2017;129:1646–1657.
- Oksenhendler E, Boutboul D, Galicier L. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8-associated lymphoproliferative disorders. Blood 2019; 133(11):1186-1190.
- van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–74.
- Dispenzieri A, Fajgenbaum DC. Overview of Castleman Disease. Blood. 2020;135(16):1353-1364.
- Fajgenbaum DC, Ruth JR, Kelleher D, et al. The collaborative network approach: a new framework to accelerate Castleman’s disease and other rare disease research. Lancet Haematol, 2016;3:e150–e152.