




la Newsletter

Letizia Roggero1, Federico Pieruzzi1,2
1Dipartimento di Medicina e Chirurgia, Università degli Studi di Milano - Bicocca; 2Clinica Nefrologica ASST-Monza, Ospedale San Gerardo
Malattia di Fabry con cardiomiopatia ipertrofica | La diagnosi di...
La diagnosi di Malattia di Fabry, nel contesto delle...
Decorso clinico
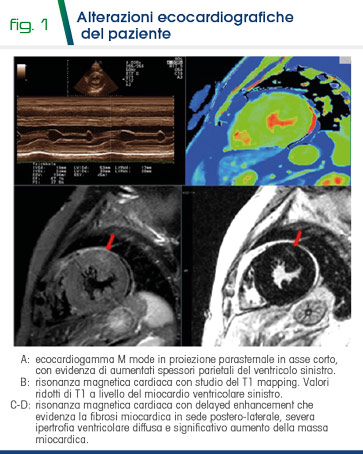 Uomo di 60 anni affetto da MF, portatore della mutazione p.N215S, variante late-onset. Nel 1994, all’età di 34 anni, ricovero ospedaliero per polmonite e pericardite, in quell’occasione evidenza di alterazioni elettrocardiografiche (IVS e onda T invertita). Dal 2004 il paziente era in follow-up cardiologico per il riscontro di cardiomiopatia ipertrofica familiare, presentava alterazioni ecocardiografiche quali ipertrofia ventricolare sinistra simmetrica (LV massa 161 g/m2) e ipertrofia ventricolare del setto (SIV 17 mm) (Fig 1).
Uomo di 60 anni affetto da MF, portatore della mutazione p.N215S, variante late-onset. Nel 1994, all’età di 34 anni, ricovero ospedaliero per polmonite e pericardite, in quell’occasione evidenza di alterazioni elettrocardiografiche (IVS e onda T invertita). Dal 2004 il paziente era in follow-up cardiologico per il riscontro di cardiomiopatia ipertrofica familiare, presentava alterazioni ecocardiografiche quali ipertrofia ventricolare sinistra simmetrica (LV massa 161 g/m2) e ipertrofia ventricolare del setto (SIV 17 mm) (Fig 1).
Nel 2008 venne posta diagnosi di MF nel fratello, anche lui in follow-up cardiologico per cardiomiopatia ipertensiva. In seguito a screening familiare, all’età di 48 anni, anche il nostro paziente è risultato positivo all’indagine genetica per MF. Il paziente, successivamente studiato, dal punto di vista sistemico presentava: un lieve interessamento nefrologico con microalbuminuria, ipoacusia neurosensoriale, acufeni e modesti disturbi gastrointestinali con alterazioni dell’alvo. Diversamente, non presentava acroparestesie, angiocheratomi, cornea verticillata, né coinvolgimento neurologico centrale.
Nel 2011 fu posto in terapia enzimatica sostitutiva (ERT) con algasidasi alfa in regime domiciliare. Con la disponibilità della terapia chaperonica orale nel 2018, su richiesta del paziente e per il progressivo peggioramento clinico definito sui parametri ecocardiografici e sull’instabilità clinica definita mediante il Fabry Stabilization Index, venne sostituita la ERT con migalastat.
Discussione
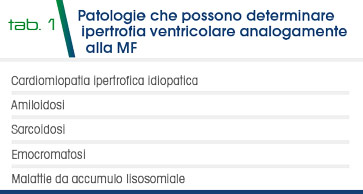 Le caratteristiche di imaging spesso non sono dirimenti nella diagnosi differenziale delle cause di cardiomiopatia ipertrofica e possono porre il clinico davanti alla necessità di escludere diverse patologie sistemiche (amiloidosi; malattie da accumulo lisosomiale, sarcoidosi, etc) (Tab.1).
Le caratteristiche di imaging spesso non sono dirimenti nella diagnosi differenziale delle cause di cardiomiopatia ipertrofica e possono porre il clinico davanti alla necessità di escludere diverse patologie sistemiche (amiloidosi; malattie da accumulo lisosomiale, sarcoidosi, etc) (Tab.1).
Le caratteristiche ecocardiografiche potevano suggerire la diagnosi di amiloidosi, esclusa, invece, dagli esami laboratoristici. Diversamente la familiarità ed il coinvolgimento sistemico potevano suggerire la MF.
La cardiomiopatia ipertrofica è un riscontro frequente nella pratica clinica; lo studio di questa patologia, dal punto di vista genetico e dell’imaging, ha permesso di rivelare come in realtà questa definizione comprenda un insieme di patologie molto eterogenee tra loro, seppur caratterizzate tutte dall’IVS. Nella MF l’età di coinvolgimento cardiaco è solitamente più tardiva rispetto alle altre forme di cardiomiopatia, ma più precoce della cardiomiopatia amiloidotica da transtiretina (2).
La familiarità, X-Linked nella MF, può guidare la diagnosi, così come l’interessamento sistemico (3). Raramente, gli esami di imaging permettono di differenziare le cause di cardiomiopatia con certezza. La risonanza magnetica cardiaca non distingue le diverse forme di cardiopatia ipertrofica sulla base della morfologia ventricolare o degli spessori parietali, ma grazie all’utilizzo del mezzo di contrasto ed alle recenti mappature disponibili, è la tecnica attualmente più idonea per porre una corretta diagnosi differenziale. Numerosi studi hanno, infatti, dimostrato una localizzazione preferenziale delle aree di “delayed enhancement” a livello della parete basale infero-laterale e posterolaterale, presente anche in assenza di franca ipertrofia ventricolare. Tale localizzazione può rappresentare un elemento di sospetto per MF nell’ambito della diagnosi differenziale in soggetti con cardiomiopatia ipertrofica idiopatica (4). Inoltre, la risonanza magnetica con mappatura T1 può suggerire un coinvolgimento cardiaco specifico di MF, in quanto il riscontro di bassi valori di T1, evidenziati solo nei pazienti affetti da MF, rifletterebbe l’accumulo di sfingolipidi nei miocardiociti, che sono associati a cambiamenti strutturali ed elettrocardiografici precoci (5).
Non bisogna trascurare l’importante contributo dell’elettrocardiogramma nella diagnosi differenziale delle cardiomiopatie ipertrofiche. Infatti, tra le alterazioni elettrocardiografiche significative, nei pazienti affetti da MF si possono rilevare voltaggi elevati con marcate anomalie della ripolarizzazione, indipendenti dalla sintomatologia clinica e dal grado di IVS. Inoltre, nel 15% dei casi si ha il riscontro di un PR corto con una conduzione atrioventricolare che, nelle fasi più avanzate, può evolvere verso blocchi atrio-ventricolari di vario grado (3).
Il coinvolgimento sistemico può aiutare nella diagnosi differenziale soprattutto nelle forme classiche di MF, per la presenza di sintomi caratteristici: acroparestesie soprattutto delle mani e dei piedi, febbri ricorrenti sine causa, disturbi gastrointestinali, anomalie della sudorazione, ipoacusia neurosensoriale ed acufeni, coinvolgimento nefrologico ed alterazioni neurologiche quali ictus e attacco ischemico transitorio (TIA). Nel caso clinico riportato, invece, la diagnosi può essere ancora più insidiosa dato che nelle forme tardive o nelle varianti cardiache di MF (come per la variante genica p.N215S), la cardiomiopatia può essere l’unica manifestazione clinica significativa.
Conclusioni
Appare evidente come la MF possa simulare la diagnosi di diverse forme di cardiomiopatia ipertrofica, inducendo a diagnosi errate o molto ritardate. Risultano di fondamentale importanza la diagnostica differenziale e soprattutto un accurato e specifico utilizzo degli esami di imaging, in quanto sono disponibili terapie specifiche in grado, se avviate precocemente, di avere un impatto favorevole sulla prognosi.
Bibliografia
- Ommen SR, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2020 Nov 20:S0735- 1097(20)36413-5.
- Maurer MS, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075.
- Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007; 93:528-35.
- De Cobelli F, Esposito A, Belloni E, et al. Delayed-enhanced cardiac MRI for differentiation of Fabry’s disease from symmetric hypertrophic cardiomyopathy. AJR Am J Roentgenol 2009;192:97- 102.
- Camporeale A, Pieroni M, Pieruzzi F, et al. Predictors of Clinical Evolution in Prehypertrophic Fabry Disease. Circulation. Cardiovascular Imaging, 12(4), e008424.