




la Newsletter
Malattia di Wilson: dal vissuto clinico del paziente la fotografia...
La malattia di Wilson (MW) è una malattia ereditaria monogenica...
La malattia di Wilson (MW) è una malattia ereditaria monogenica autosomica recessiva dovuta alla mutazione del gene che codifica per una proteina l’ATPasi7B, che regola il trasporto del rame all’interno della cellula epatica [1].
Le mutazioni di questo gene causano un deficit di escrezione di rame nella bile, la principale via di eliminazione del metallo dall’organismo. Ne consegue un accumulo nel fegato, dove può causare una malattia che può manifestarsi con un ampio spettro clinico che va dalla steatoepatite alla cirrosi o all’insufficienza epatica acuta e successivamente in tutti gli organi e tessuti, in particolare nel cervello, nella cornea, nel rene e nel cuore.
Ciò rende ragione del fenotipo variabile della malattia che peraltro si caratterizza nella maggior parte dei casi con presentazione epatica associata o meno ad alterazioni neurologiche o sintomi psichiatrici. Si stima che la prevalenza della MW in Italia sia pari a 1:30.000 ma in alcune regioni come in Sicilia, Puglia, Campania e soprattutto in Sardegna è decisamente maggiore [2,3].
La rarità della malattia e l’eterogeneità dell’espressività clinica fanno sì che la malattia non venga riconosciuta e che la diagnosi e l’inizio del trattamento vengano ritardati.
Opzioni terapeutiche
La MW se non trattata, ha sempre esito fatale ma è anche una delle poche malattie ereditarie monogeniche per la quale sono disponibili farmaci efficaci quali, la penicillamina, la trientina e i sali di zinco, in grado di arrestare la progressione della malattia in più dell’80% dei pazienti e di consentire, in questi soggetti, una aspettativa di vita paragonabile a quella della popolazione generale [4].
Il trattamento deve però essere proseguito per tutta la vita; infatti, la sua interruzione porta inevitabilmente a morte per insufficienza epatica. Il riconoscimento precoce e l’aderenza alla terapia sono cruciali nel determinare la prognosi della malattia.
La survey
Sulla base di queste considerazioni in collaborazione con l’Associazione Nazionale della malattia di Wilson è stata condotta un’indagine conoscitiva sulle caratteristiche demografiche e cliniche, sul percorso diagnostico e terapeutico, sulla qualità di vita e sul vissuto di malattia dei pazienti italiani.
L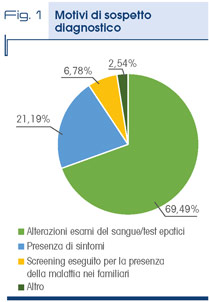 a survey, di natura qualitativa, è stata condotta su un campione di 151 pazienti, di questi 121 analizzabili: il 52% erano donne e il 69,3% di età superiore a 18 anni. I dati che emergono sono interessanti soprattutto per quanto riguarda i tempi di diagnosi. I casi pediatrici sono diagnosticati entro 12 mesi dalla comparsa dei sintomi, mentre per i pazienti adulti il ritardo diagnostico varia dai 2-5 anni. La diagnosi precoce nei bambini è certamente facilitata dallo screening familiare, in seguito alla diagnosi di malattia di un familiare.
a survey, di natura qualitativa, è stata condotta su un campione di 151 pazienti, di questi 121 analizzabili: il 52% erano donne e il 69,3% di età superiore a 18 anni. I dati che emergono sono interessanti soprattutto per quanto riguarda i tempi di diagnosi. I casi pediatrici sono diagnosticati entro 12 mesi dalla comparsa dei sintomi, mentre per i pazienti adulti il ritardo diagnostico varia dai 2-5 anni. La diagnosi precoce nei bambini è certamente facilitata dallo screening familiare, in seguito alla diagnosi di malattia di un familiare.
La diagnosi è stata posta nella maggior parte dei casi dal gastroenterologo, e dal pediatra nei pazienti più giovani. In circa il 15% dei casi i pazienti hanno attribuito la diagnosi al genetista. È altresì verosimile che in questi casi la diagnosi sia stata sospettata da altri specialisti che abbiano dato indicazione ad effettuare l’analisi genetica.
Nel 7% dei casi la diagnosi è avvenuta in seguito alla esecuzione del test genetico nei familiari di pazienti con la malattia (Fig. 1).
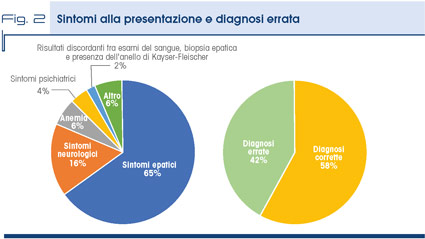 Il motivo iatrotropico è rappresentato generalmente dal riscontro, spesso occasionale, di un’alterazione dei test epatici, in particolare delle transaminasi, ma in una quota consistente dei casi da sintomi neurologici e psichiatrici che in qualche modo avrebbero potuto far sospettare la MW: nonostante ciò è stata formulata una diagnosi errata nel 42% dei casi (Fig. 2).
Il motivo iatrotropico è rappresentato generalmente dal riscontro, spesso occasionale, di un’alterazione dei test epatici, in particolare delle transaminasi, ma in una quota consistente dei casi da sintomi neurologici e psichiatrici che in qualche modo avrebbero potuto far sospettare la MW: nonostante ciò è stata formulata una diagnosi errata nel 42% dei casi (Fig. 2).
Questo dato rafforza l’ipotesi che il problema sia la mancata conoscenza o il mancato sospetto di malattia.
La penicillamina è risultato essere il trattamento iniziale per il 66% dei pazienti mentre lo zinco per il 22% dei casi; una minima quota ha avuto come trattamento iniziale la trientina o la combinazione chelante + zinco.
In particolare, la penicillamina viene utilizzata nel trattamento del paziente adulto che generalmente ha una malattia di fegato più avanzata, mentre lo zinco nei giovani sotto i 18 anni, in cui alla diagnosi è presente una malattia più lieve quale la steatoepatite.
In questa fascia sono collocati per lo più i pazienti pre-sintomatici, per i quali lo zinco rappresenta il farmaco di prima scelta. L’impiego così limitato della trientina non deve sorprendere, in quanto questo farmaco ha indicazione per il trattamento dei pazienti intolleranti alla penicillamina.
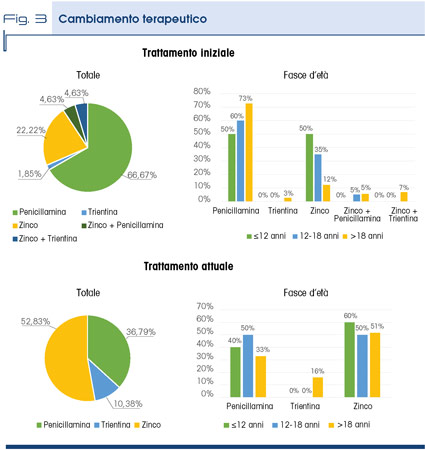 Se si analizza la risposta dei pazienti relativa al trattamento in corso al momento della survey si può osservare che la quota di pazienti in penicillamina è quasi dimezzata con un corrispondente aumento della frazione dei pazienti trattati con trientina e zinco. Il cambiamento di terapia è attribuibile non solo al fatto che la terapia iniziale non abbia ottenuto la risposta attesa ma soprattutto al fatto che i pazienti trattati con penicillamina in una percentuale rilevante di casi hanno presentato degli effetti collaterali (Fig. 3).
Se si analizza la risposta dei pazienti relativa al trattamento in corso al momento della survey si può osservare che la quota di pazienti in penicillamina è quasi dimezzata con un corrispondente aumento della frazione dei pazienti trattati con trientina e zinco. Il cambiamento di terapia è attribuibile non solo al fatto che la terapia iniziale non abbia ottenuto la risposta attesa ma soprattutto al fatto che i pazienti trattati con penicillamina in una percentuale rilevante di casi hanno presentato degli effetti collaterali (Fig. 3).
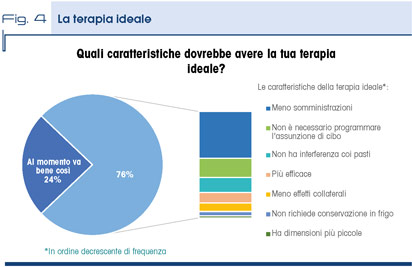
Il 76% dei pazienti non è soddisfatto della terapia in corso; i pazienti riportano che la terapia ideale oltre che essere più efficace dovrebbe avere meno effetti collaterali, un minor numero di capsule/compresse da assumere e presentare minori difficoltà nella assunzione della dose prescritta (Fig. 4).
I risultati
 Da questa fotografia del “mondo” dei pazienti con MW emerge, da una parte, la necessità di una maggiore formazione e sensibilizzazione dei medici specialisti e medici di medicina generale e, dall’altra, di continuare la ricerca di nuovi farmaci o formulazioni farmaceutiche in grado di favorire l’aderenza e la soddisfazione per la terapia.
Da questa fotografia del “mondo” dei pazienti con MW emerge, da una parte, la necessità di una maggiore formazione e sensibilizzazione dei medici specialisti e medici di medicina generale e, dall’altra, di continuare la ricerca di nuovi farmaci o formulazioni farmaceutiche in grado di favorire l’aderenza e la soddisfazione per la terapia.
La recente disponibilità in Italia della trientina tetracloridrato costituisce un passo rilevante in questa direzione.
Ringraziamenti: ai pazienti e ai loro familiari per la partecipazione; al Comitato Scientifico dell'Associazione Pazienti WD per la discussione dei risultati; alla dott.ssa Carnevali e all'Azienda GMP-Orphan per il supporto incondizionato al progetto.
Bibliografia
- Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 1993;5:344-50.
- Ala A, Walker AP, Ashkan K, et al. Wilson’s disease. Lancet 2007;369:397-408.
- Prevalenza o numero dei casi pubblicati elencati in ordine alfabetico delle malattie Gennaio 2020- n°1 – Orphanet
https://www.orpha.net/orphacom/cahiers/docs/IT/Prevalenza_delle_malattie_rare_in_ordine_alfabetico.pdf (ultimo accesso 07/04/2020) - Bruha R, Marecek L, Pospisilova L, et al. Long-term follow-up of Wilson Disease: natural history, treatment, mutation analysis and phenotypic correlation. Liver Int 2011;31:83-91.