




la Newsletter
Malattie rare non diagnosticate | La medicina genomica può essere...
La medicina genomica può essere usata per la diagnosi di malattie...
Genomic medicine for undiagnosed diseases
Wise AL, Manolio TA, Mensah GA, et al. Lancet 2019; 394: 533–40.
Riassunto
Questo è il terzo di una serie di 5 articoli che hanno lo scopo di introdurre il medico clinico alla comprensione delle opportunità diagnostiche offerte dalla medicina genomica alla pratica clinica.
L’articolo che qui commentiamo discute come la medicina genomica possa essere usata per la diagnosi di malattie rare che non hanno ancora ottenuto una diagnosi con i metodi tradizionali e come possa modificare il percorso terapeutico (Fig. 1).
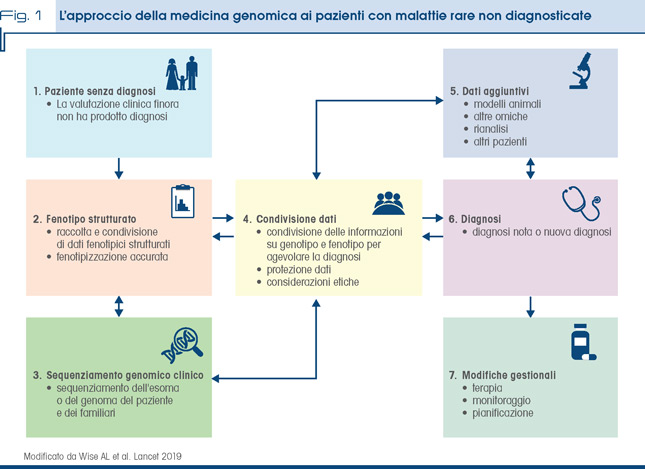
I tempi sono maturi per una entrata più massiccia nella pratica clinica: il costo di exome e genome sequencing si sta sempre più riducendo ed è ben chiara in ambito medico-scientifico la necessità di migliorare i tempi diagnostici delle malattie rare.
Molti lavori pubblicati negli ultimi anni dimostrano l’efficacia del sequenziamento genomico associato ad una accurata definizione del fenotipo utilizzando strumenti standardizzati.
Esistono strumenti elettronici che aiutano nella definizione strutturata del fenotipo: Phenomizer classifica le malattie sulla base di segni e sintomi e produce una diagnosi differenziale basata sul fenotipo, BioLark si può usare per produrre termini human phenotype ontology (HPO) (sistema di definizione standardizzata di termini clinici) da note cliniche, Phenolyzer combina i fenotipi con precedenti conoscenze biologiche per risalire a possibili geni implicati.
Il sequenziamento genomico associato a raccolta strutturata dei dati fenotipici ha permesso di raggiungere più velocemente la diagnosi in neonati con malattia acuta o in feti con anomalie morfologiche, rispetto ai metodi tradizionali, con possibilità di intervento terapeutico più mirato, prognosi più precoce e risparmio economico. In questo ambito è importante anche la condivisione dei dati fenotipici nelle malattie rare, sia dei feti che dei pazienti, per confermare l’associazione genotipo/fenotipo, per identificare altri pazienti con stesso fenotipo, per rinforzare l’evidenza che una variante di un gene è associata ad un certo fenotipo.
Esiste ormai una letteratura che mostra come, a scopo diagnostico, i dati fenotipici e quelli genomici possono essere integrati anche con dati da modelli animali, da metaboloma e transcrittoma.
La frequenza diagnostica del clinical exome sequencing è molto variabile ma possiamo dire che, in pazienti già esaminati senza successo con metodi tradizionali, si ha una frequenza complessiva media di 25-35% con una percentuale più bassa negli adulti.
In molti casi ciò ha portato a modificare procedure terapeutiche (ad esempio diverso approccio ad intervento in feto per ernia diaframmatica, in osteogenesi imperfetta programmazione di parto senza traumi e apprendimento prima del parto da parte dei genitori di come evitare traumi al bambino) o a evitare inutili stress e a programmare cure palliative.
Tre articoli scientifici mostrano come la diagnosi genomica veloce in terapia intensiva neonatale abbia modificato la strategia terapeutica nel 33-72% dei casi.
È ancora da discutere in comunità scientifica come riportare al paziente o alla famiglia tutti i risultati secondari che si ottengono con il sequenziamento genomico.
Commento
Questo articolo, che spiega ai clinici come potrebbero utilizzare al meglio il sequenziamento genomico, apre nuovi orizzonti che devono essere esplorati.
Il clinico deve imparare a raccogliere in modo strutturato i dati fenotipici usando programmi disponibili sul web e a condividerli.
A tal proposito vediamo qualche difficoltà per il momento: 1- in questo articolo si insiste molto sui vantaggi del sequenziamento genomico veloce nella diagnosi di neonati con malattia acuta o di feti, ma non ci risulta che per il momento questo sia possibile in Italia salvo che in situazioni molto particolari. 2- l’uso più esteso del sequenziamento genomico nella diagnosi delle malattie rare impone la risoluzione di vari problemi etici quali, per primo, quello degli incidental finding: quanto delle informazioni genomiche secondarie va riportato al paziente o alla sua famiglia?
Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease
Kim J, Hu C, Moufawad El Achkar C, et al. N Engl J Med. 2019;381(17):1644-1652.
Riassunto
Il sequenziamento del genoma è spesso fondamentale nella caratterizzazione delle malattie rare, tuttavia per la maggior parte delle patologie su base genetica oggi diagnosticabili non è ancora disponibile un trattamento mirato.
Il lavoro espone come, partendo dalla caratterizzazione molecolare di una rara malattia neurodegenerativa a prognosi infausta, si sia arrivati alla progettazione, alla valutazione preclinica e alla produzione di un farmaco specifico.
La piccola paziente descritta è affetta da Ceroidolipofuscinosi neuronale tipo 7 (nota anche come malattia di Batten o CLN7), condizione che si manifesta con un deterioramento della vista nei primi anni di vita. Dopo l'esordio dei sintomi visivi iniziano il declino cognitivo e l'epilessia. La cecità sopraggiunge entro pochi anni e contestualmente le capacità cognitive e motorie si deteriorano progressivamente. Il trattamento è solo sintomatico e consiste in cure palliative con la somministrazione di farmaci anticonvulsivanti e in interventi di supporto.
Le Ceroidolipofuscinosi sono malattie autosomiche recessive e sono causate dal difetto/mutazione di un singolo gene (monogeniche). Ciascun gene è costituito da due alleli: un allele viene ereditato dalla madre, l'altro dal padre. Affinché la malattia si manifesti, è necessario che l'individuo erediti l'allele recessivo mutato da entrambi i genitori che sono portatori della malattia. Grazie a sofisticate indagini genetiche, è stato possibile identificare il difetto responsabile della malattia nella piccola paziente: una mutazione patogenetica nota in una copia del gene CLN7 e, nell'altra copia, una anomalia molto complessa e non descritta in precedenza condizionante errori di sintesi dell'RNA messaggero (mRNA).
Gli autori hanno ideato oligonucleotidi antisenso (antisense oligonucleotide, ASO) con l’obiettivo di "correggere" il difetto dell'mRNA e hanno selezionato un ASO, dimostratosi in grado di indurre un aumento del rapporto tra mRNA normale e mutante nei fibroblasti in coltura della paziente.
Dopo una valutazione tossicologica abbreviata, e dopo aver ottenuto l'autorizzazione dalla Food and Drug Administration (FDA) e l'approvazione dal comitato etico competente, i ricercatori hanno somministrato il composto alla piccola per via intratecale a dosi crescenti. Il trattamento, iniziato all’età di sei anni, si è dimostrato in grado di influenzare positivamente il decorso della malattia, in assenza di gravi effetti collaterali.
Commento
Nel caso descritto la diagnostica e la caratterizzazione molecolare hanno reso possibile la sintesi di un oligonucleotide antisenso (ASO) specifico per la correzione del difetto studiato. Gli ASO sono brevi molecole a singolo filamento di DNA o di RNA complementari ad una determinata sequenza. La sequenza a cui si legano è solitamente una molecola di mRNA che, “allacciata” da un ASO specifico, non è più in grado di essere tradotta. Le ricerche sperimentali condotte sulle linee cellulari ottenute dalla paziente hanno aperto la strada alla realizzazione di uno studio clinico su base individuale (N-of-1 trial) entro un anno.
Lo studio, finanziato in larga parte da una Fondazione creata dai familiari della piccola paziente (Mila’s Miracle Foundation) rappresenta un modello per il celere sviluppo di terapie personalizzate. I tempi di realizzazione del trattamento sono rapidissimi se confrontati con quelli abitualmente necessari per portare un principio attivo dalla sua identificazione come potenziale farmaco alla pratica clinica (Fig. 2).
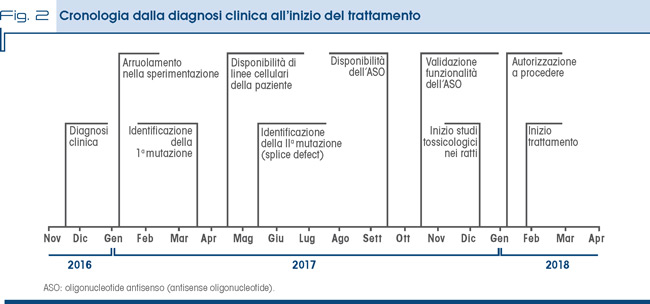
Il lavoro illustra come la definizione delle basi genetiche possa farci comprendere i meccanismi responsabili di determinati fenotipi di malattie rare e come le tecnologie recentemente sviluppate consentano l’avvio di percorsi per lo sviluppo di farmaci davvero individualizzati.
Sebbene questo nuovo percorso per la scoperta e lo sviluppo di farmaci sia più avanzato per gli ASO, altri tipi di trattamenti, tra cui terapie cellulari e genetiche individualizzate, si stanno concretamente avvicinando.
Le possibilità di realizzazione di terapie sofisticate e molto costose aprono nuovi interrogativi. In che modo l'urgenza della situazione del paziente o il numero di persone che alla fine potrebbero essere curate influenzeranno il processo decisionale? Se le possibilità di interventi individualizzati diventeranno comuni, dovranno essere affrontati anche gli aspetti relativi all'approvazione e alla sostenibilità dei trattamenti.
Nell’immediato futuro tali tematiche saranno sempre più oggetto di discussione da parte dalle autorità regolatorie e sarà fondamentale il contributo di accademici, associazioni dei pazienti, industria farmaceutica e altre parti interessate.