




la Newsletter

Silvia Scupilliti1,2, Angelo Selicorni1
1UOC Pediatria. Presidio S. Fermo. ASST-Lariana, Como. Centro Fondazione Mariani per il Bambino Fragile; 2Università degli Studi di Milano - Scuola di Specializzazione in Genetica Medica
Mutazioni nei geni COL4A1-A2 | Varianti patogenetiche nei geni...
Varianti patogenetiche nei geni COL4A1 e COL4A2 sono responsabili...
Varianti patogenetiche nei geni COL4A1 e COL4A2 sono alla base di malattie sistemiche rare che predispongono ad emorragia intracranica e al coinvolgimento vascolare a livello renale, oculare, cardiaco e muscolare. L’emorragia intracranica fetale rappresenta un evento raro con una prevalenza stimata di 1:10.000 gravidanze, associato a un significativo rischio di conseguenze neurologiche a lungo termine e morte fetale nei casi più gravi; può manifestarsi come porencefalia, leucoencefalia e aneurismi intracranici. Nella maggior parte dei casi la causa di emorragia intracranica fetale rimane inspiegabile (1).
In passato la presenza di porencefalia era stata spesso considerata il risultato di un insulto esterno, ad esempio sanguinamento perinatale e post-anossico senza alcuna base genetica e in assenza di una coagulopatia.
Solo all’inizio degli anni 2000 è stata identificata la presenza di mutazioni nei geni COL4A1 e COL4A2 come causa di porencefalia, a trasmissione autosomica dominante (2).
I geni COL4A1 e COL4A2 codificano rispettivamente per le catene a1 e a2 di collagene di tipo IV. Il collagene di tipo IV è un componente del collagene non fibrillare, costituente principale delle membrane basali di molti tessuti, tra cui quella dell’endotelio vascolare (3).
Caratteristiche cliniche
Le conseguenze cliniche di una mutazione del gene COL4A1 sono estremamente variabili per gravità, tipologia ed età di insorgenza con ampie variazioni intra- ed interfamiliari (Tab. 1).

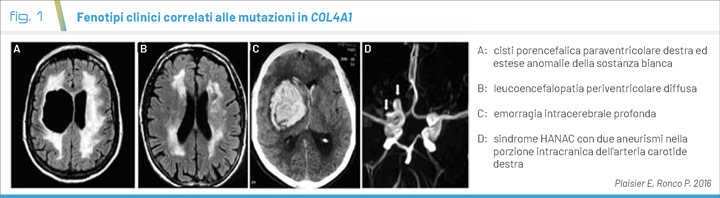
Alcune persone infatti sono del tutto asintomatiche mentre altre possono sviluppare complicanze gravi, anche potenzialmente letali. Alcuni soggetti possono sviluppare solo sintomi specifici come emicranie isolate o ictus durante l'infanzia o l'età adulta. Il numero ridotto di pazienti ad oggi identificati non permette di definire un quadro completo della variabilità clinica e prognostica. Esiste infatti un ampio spettro di fenotipi clinici correlati alle mutazioni in COL4A1 (OMIM 120130) (Fig. 1) (4):
- porencefalia familiare autosomica dominante di tipo 1 (OMIM 175780): “porencefalia” è un termine usato per qualsiasi cavitazione o cisti piena di liquido cerebrospinale nell’encefalo. Le cavità porencefaliche (Fig. 1A) derivano dalla rottura delle membrane basali cerebrovascolari. Sono uno dei reperti più frequentemente identificati nei pazienti con mutazione in COL4A1.
Oltre alle cavità porencefaliche, l'imaging cerebrale può mostrare vari gradi di leucoencefalopatia periventricolare, microsanguinamenti, infarto lacunare, ventricoli dilatati e calcificazioni. L'aumentata fragilità di questi vasi li rende suscettibili all'emorragia, già in utero o per traumi alla nascita, anche se il rischio rimane per tutta la vita. - microangiopatia e leucoencefalopatia pontina autosomica dominante (PADMAL, OMIM 618564): malattia dei piccoli vasi che provoca l'insorgenza di ictus ischemici ricorrenti; differisce dalla porencefalia familiare autosomica dominante per l'assenza di cavità porencefaliche.
L'imaging cerebrale mostra il coinvolgimento caratteristico dei piccoli vasi cerebrali soprattutto del distretto pontino, tra cui leucoencefalopatia periventricolare diffusa (Fig. 1B), infarti lacunari e calcificazioni intracerebrali.
Le persone affette sviluppano compromissione cognitiva e motoria progressiva, ma variabile, coerente con la demenza progressiva multi-infartuale. - sindrome da angiopatia ereditaria con nefropatia, aneurismi e crampi muscolari (sindrome HANAC, OMIM 611773): è un quadro clinico multi sintomatico sembra secondario a mutazioni in COL4A1 che colpiscono i residui di glicina in prossimità degli esoni 24 e 25 (2). Il coinvolgimento cerebrale è rappresentato da aneurismi intracranici singoli o multipli (Fig. 1D), tutti localizzati sul sifone carotideo; la metà degli individui affetti presenta malattia dei piccoli vasi cerebrali; nessuno ha porencefalia. Il coinvolgimento renale si esprime con ematuria microscopica isolata ed episodi intermittenti di ematuria macroscopica; possibile la presenza di cisti renali corticali e midollari bilaterali.
È descritto un coinvolgimento muscolare con un aumento persistente della concentrazione sierica di CK e crampi muscolari. La tortuosità arteriolare retinica bilaterale è osservata in tutti i pazienti con sindrome HANAC. Altre manifestazioni possono comprendere il fenomeno di Raynaud, l'aritmia sopraventricolare e cisti epatiche.
Le mutazioni nel gene COL4A2 (OMIM 120090), invece, sono responsabili della porencefalia autosomica dominante di tipo 2 (presentazione simile al tipo 1): malattia dei piccoli vasi con compromissione neurologica variabile derivante da un'irrorazione vascolare disturbata che porta alla degenerazione cerebrale, si manifesta all’imaging come porencefalia. Le persone affette presentano tipicamente emiplegia, convulsioni e disabilità intellettiva, sebbene la gravità sia variabile.
Diagnosi
A causa dell'espressione clinica estremamente variabile con ampie variazioni intra- ed interfamiliari ed evidenza di penetranza incompleta (5), le mutazioni dei geni del collagene di tipo IV sono spesso sotto diagnosticate. Il sequenziamento dei geni COL4A1 e COL4A2 dovrebbe essere preso in considerazione in tutti i neonati con una cisti porencefalica e/o emiparesi alla nascita così come nei neonati con emorragia intracranica di eziologia sconosciuta.
Recentemente, il test genetico è stato raccomandato anche nei neonati con malattia emolitica di eziologia sconosciuta, specialmente quando vengono identificate altre manifestazioni correlate a COL4A1 (6). Tra i meccanismi patogenetici ad oggi non sono state riportate delezioni o duplicazioni intrageniche ma, quasi esclusivamente, varianti missenso. In relazione a ciò una ricerca di duplicazioni/delezioni di grandi dimensioni può avere una resa molto bassa (6).
Genetica
I disturbi correlati a COL4A1 e a COL4A2 sono ereditati in modo autosomico dominante e coinvolgono maschi e femmine in numero uguale. Almeno il 50% delle persone con diagnosi di disturbo correlato a COL4A1 ha un genitore affetto. Si stima che il 27% dei pazienti abbia una variante insorta de novo. La penetranza dei disturbi correlati a COL4A1 è incompleta, per alcuni autori invece è prossima al 100% (4), con un'espressività variabile per età di insorgenza e gravità dei sintomi clinici, anche nella stessa famiglia.
Diagnosi differenziale
Ci sono alcune condizioni che possono simulare i disturbi correlati a COL4A1 e COL4A2:
- Arteriopatia cerebrale autosomica dominante con infarti subcorticali e leucoencefalopatia (CADASIL, OMIM 125310): è causata dalla mutazione in eterozigosi nel gene NOTCH3. CADASIL è un disturbo progressivo dei piccoli vasi arteriosi del cervello che si manifesta con cefalea migrante con aura, ictus e lesioni della sostanza bianca con conseguente deterioramento cognitivo. Il segno patognomonico è la presenza di granuli densi nella tonaca media delle arteriole che possono essere identificati al microscopio elettronico dopo biopsia cutanea.
- Vasculopatia retinica autosomica dominante con leucodistrofia cerebrale (RVCL, OMIM 192315): è causata dalla mutazione in eterozigosi del gene TREX1. È una malattia autosomica dominante che insorge in età adulta e che coinvolge i piccoli vasi encefalici provocando la degenerazione del sistema nervoso centrale con progressiva perdita della vista, ictus, compromissione motoria e declino cognitivo. Un sottotipo di RVCL è l’HERNS (endoteliopatia con retinopatia, nefropatia e ictus), oltre il distretto cerebrale coinvolge la membrana basale gromerulare e di altri tessuti.
- Le cisti porencefaliche possono essere conseguenza di un infarto emorragico parenchimale prenatale o neonatale nel contesto della trombocitopenia alloimmune neonatale o a causa di coagulopatie come la malattia di von Willebrand (OMIM 613160), il deficit del fattore V (OMIM 227400) o il deficit del fattore X (OMIM 227600).
- Arteriopatia cerebrale autosomico recessiva con infarti sottocorticali e leucoencefalia (CARASIL o sindrome di Maeda, OMIM 600142): è un'arteriopatia cerebrale dei piccoli vasi non ipertensiva, caratterizzata da alopecia, spondilosi, disfunzione motoria progressiva e demenza. L'esordio è solitamente nella seconda o terza decade di vita con disturbo dell’andatura da spasticità. La CARASIL è causata da varianti patogene nel gene HTRA1. Il 23% delle persone interessate da questa malattia presenta episodi simili a ictus prima dei 40 anni.
- Anoftalmia/microftalmia (A/M): è una malattia geneticamente eterogenea che è stata associata a varianti patogene in più di 70 geni. SOX2, OTX2 e FOXE3 sono tra i geni più comunemente associati ad A/M.
Gestione clinica
Nelle gravidanze in cui il feto ha un rischio di disturbo correlato a COL4A1 è raccomandato il parto cesareo per prevenire lesioni vascolari attribuibili ai traumi del parto.
Non esistono protocolli di trattamento standardizzati o linee guida per le persone affette. Le terapie si basano sui sintomi specifici di ogni individuo. Ragionevole è una valutazione clinica annuale, può essere proposto un imaging cerebrale regolare, in particolare per valutare la dimensione degli aneurismi cerebrali asintomatici.
Importante la prevenzione delle manifestazioni primarie: evitare l’esposizione agli anticoaugulanti e alle attività che comportano un aumentato rischio di trauma cranico per ridurre il rischio di emorragia intracranica; evitare il fumo e ridurre l’ipertensione per un rischio aumentato di ictus.
Dopo la diagnosi iniziale, per stabilire l'estensione della malattia si raccomanda l’esecuzione di: RMN cerebrale; TAC angiografica cerebrale; valutazione oftalmologica con valutazione del fundus oculi ed esame con lampada a fessura; ecografia o TAC addominale; misurazione della concentrazione sierica di CK; misurazione della concentrazione di creatinina sierica e stima della velocità di filtrazione glomerulare; valutazione per la presenza di ematuria; elettrocardiogramma (ECG) ed ecocardiografia.
Conclusioni
In alcuni pazienti con storia prenatale e perinatale complicata, le malformazioni cerebrali possono essere erroneamente diagnosticate come conseguenza diretta dell'insulto pre/perinatale e i test genetici possono anche non essere raccomandati. L’esistenza dei disturbi correlati a COL4A1 (e COL4A2) suggerisce l'importanza della valutazione genetica e di una diagnosi precoce anche nei pazienti con causa apparentemente "non genetica" di malformazioni congenite come la porencefalia e specialmente nei casi di sanguinamento intracerebrale fetale altrimenti inspiegabili (1).
Bibliografia
- Straka B, Vlčková M, Libá Z, et al. COL4A1 mutation‐related disorder presenting as fetal intracranial bleeding, hydrocephalus, and polymicrogyria. Epilepsia Open. 2023;8:211–216.
- Meuwissen MEC, Halley DJJ, Smit LS, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med. 2015;17 (11): 843–53.
- Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet.2012; 21 (R1): R97–110.
- Plaisier E, Ronco P. COL4A1-Related Disorders. 2009 Jun 25 [updated 2016 Jul 7]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. PMID: 20301768.
- Giorgio E, Vaula G, Bosco G, et al. Two families with novel missense mutations in COL4A1: When diagnosis can be missed. J Neurol Sci. 2015;352(1-2):99-104.
- Tan AP, Svrckova P, Cowan F, et al. Intracranial hemorrhage in neonates: A review of etiologies, patterns and predicted clinical outcomes. Eur J Paediatr Neurol. 2018;22 (4): 690–717.