




la Newsletter
Novità in tema di alfa-Mannosidosi | Recentemente l'EMA ha...
Recentemente l'EMA ha approvato velmanase alfa, primo enzima...
Aspetti di fisiopatologia
L’alfa-Mannosidosi (MIM 248500) è una malattia da accumulo lisosomiale causata da un deficit dell’enzima alfa-D-mannosidasi lisosomiale (EC 3.2.1.24), che partecipa alla catena di degradazione intralisosomiale delle glicoproteine. La malattia è trasmessa con modalità autosomico recessiva e rientra nel gruppo delle malattie ultra-rare, con una prevalenza di circa 1:500.000-1.000.000 nati vivi/anno (1, 2).
L’alfa-mannosidasi catalizza l’idrolisi dei legami mannosidici alfa(1,2)-, alfa(1–3)-, e alfa(1–6)- presenti negli oligosaccaridi N-linked. Il blocco della degradazione glicoproteica provoca un accumulo intralisosomiale di oligosaccaridi solubili ricchi in mannosio nelle cellule di tutti i tessuti, con conseguente danno tossico e apoptosi (3).
L’enzima è espresso in tutti i tessuti umani dove è presente in due forme isoenzimatiche denominate A e B, entrambe codificate dallo stesso gene MAN2B1 localizzato sul cromosoma 19 (19 p13.2-q12) che si compone di 24 esoni e codifica una proteina composta da 1011 aminoacidi (4). Sono state descritte mutazioni missenso, non-senso, frameshift, piccole inserzioni, duplicazioni, delezioni, mutazioni introniche di splicing e larghe delezioni.
La maggior parte delle mutazioni sono familiari, anche se 3 mutazioni comprendono circa il 30% dei casi: c.2248C>T (27%), c.1830+1G>C (5%) e c.2426 T>C (3%) (5). Ad oggi non vi è evidenza di una correlazione genotipo-fenotipo, anche all’interno della stessa famiglia (1).
Aspetti clinici
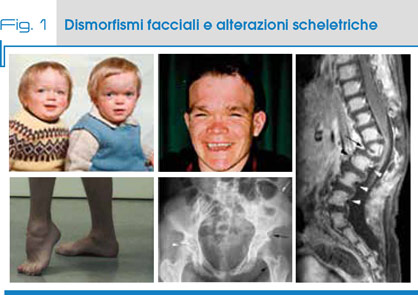 Come altre patologie lisosomiali, anche l'alfa-Mannosidosi presenta un’ampia variabilità fenotipica che varia da forme molto severe, feto-neonatali, a quadri con sintomatologia lieve. Al di fuori dalle forme ad espressione feto-neonatale, i pazienti sono in genere asintomatici alla nascita e durante il primo anno di vita, manifestando poi il progressivo comparire dei segni e sintomi caratteristici: immunodeficienza, dismorfismi facciali e scheletrici (Fig.1), ipoacusia e sordità, deficit neurologici (atassia e ipotonia muscolare), cognitivi (deficit graduale delle funzioni mentali) e del linguaggio e disturbi psichiatrici (psicosi) (Tab. 1). Possono essere presenti anche epatosplenomegalia, opacità corneale, miopatia metabolica e artrite.
Come altre patologie lisosomiali, anche l'alfa-Mannosidosi presenta un’ampia variabilità fenotipica che varia da forme molto severe, feto-neonatali, a quadri con sintomatologia lieve. Al di fuori dalle forme ad espressione feto-neonatale, i pazienti sono in genere asintomatici alla nascita e durante il primo anno di vita, manifestando poi il progressivo comparire dei segni e sintomi caratteristici: immunodeficienza, dismorfismi facciali e scheletrici (Fig.1), ipoacusia e sordità, deficit neurologici (atassia e ipotonia muscolare), cognitivi (deficit graduale delle funzioni mentali) e del linguaggio e disturbi psichiatrici (psicosi) (Tab. 1). Possono essere presenti anche epatosplenomegalia, opacità corneale, miopatia metabolica e artrite.
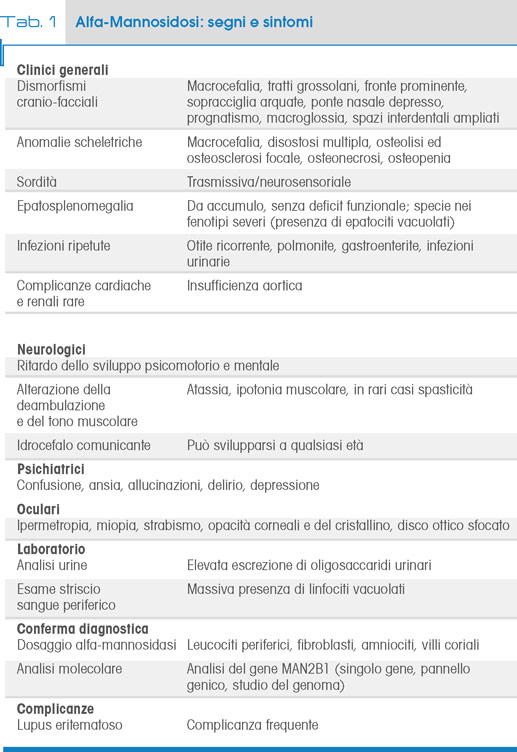
L'immunodeficienza si manifesta con infezioni ricorrenti, specialmente nella prima decade di vita, mentre gli aspetti dismorfici includono una facies con tratti grossolani (fronte prominente, sopracciglia arrotondate, sella nasale piatta, macroglossia, denti distanziati e prognatismo) e anomalie scheletriche (macrocrania, disostosi multipla che varia da leggera a moderata, scoliosi e deformazione dello sterno, artopatia). È comune un lieve strabismo.
Una classificazione recente, indirizzata a fornire una distinzione sulla base della severità clinica, ha identificato 3 fenotipi principali:
- forma lieve (tipo 1), diagnosticata dopo i dieci anni di vita, senza anomalie scheletriche e miopatia, con progressione lenta;
- forma moderata (tipo 2), con diagnosi prima dei dieci anni e presenza di interessamento scheletrico e miopatia a progressione lenta;
- forma severa (tipo 3), presente alla nascita o in epoca prenatale con exitus rapido ed interessamento neurologico e del sistema immunitario.
La variabilità clinica è significativa a testimonianza dell’esistenza di un continuum fenotipico, similmente ad altre patologie lisosomiali.
Diagnosi
Al di là degli aspetti clinici, che fanno generalmente sospettare di essere in presenza di una malattia da accumulo, nell’ambito del gruppo delle mucopolisaccaridosi o simili (Tab. 2), la conferma diagnostica si basa sulla misura dell’attività enzimatica sui leucociti o fibroblasti, mediante metodi basati su substrati colorimetrici o fluorimetrici.
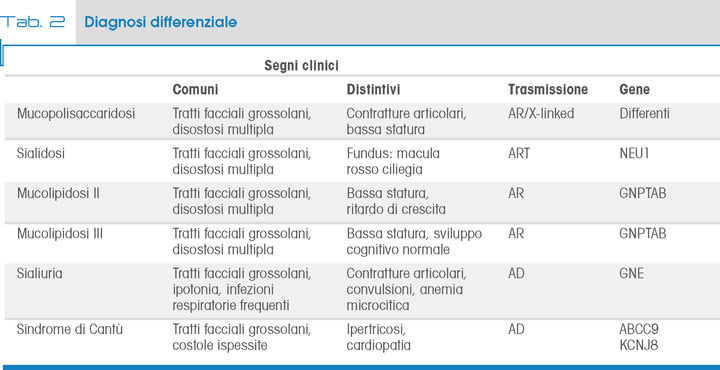
Alcuni pazienti affetti da alfa-Mannosidosi mantengono un’attività residua del 5-15%, molto probabilmente dovuta ad altra alfa-mannosidasi (l’alfa-mannosidasi presente nell’apparato del Golgi o quella citosolica).
È indicativo, anche se non diagnostico, l'aumento della secrezione urinaria di oligosaccaridi ricchi in mannosio.
L’analisi molecolare, finora eseguita a completamento dell’iter diagnostico, va sostituendo con frequenza sempre maggiore il test biochimico come approccio di prima istanza.
L’analisi prevede la sequenza dei 24 esoni presenti nel gene MAN2B1 e ha permesso ad oggi di identificare 155 varianti da 191 pazienti, che sono state raccolte nel database Amamutdb.no (http://amamutdb.no). La maggior parte dei pazienti diagnosticati appartiene al tipo 2.
La diagnosi pre-natale è possibile in presenza di un caso indice in famiglia e può essere eseguita mediante il test biochimico o l’analisi genetica della mutazione familiare su villi coriali o amniociti. In presenza della nascita di un nuovo caso, è essenziale fornire alla famiglia una consulenza genetica appropriata che aiuti a comprendere i tempi e le necessità della malattia.
Terapia
La terapia dell’alfa-Mannosidosi è stata per lungo tempo una terapia di supporto, prevalentemente rivolta a prevenire o ritardare le complicanze, che fanno parte della sua storia naturale, e a ottimizzare la qualità di vita dei pazienti.
Elementi cardine erano e sono tutt’ora: il trattamento antibiotico delle infezioni, il posizionamento del drenaggio timpanico, l’utilizzo di apparecchi acustici, la correzione dei deficit visivi, il trattamento delle complicanze ortopediche, l’uso della sedia a rotelle, il trattamento dell’idrocefalo.
Importanti sono anche gli approcci di logopedia ed educativi precoci, per mantenere e sviluppare le capacità sociali, e fisiatrici per migliorare le funzioni articolari e muscolari.
Programmare un follow-up clinico e funzionale annuale, o con frequenza più ravvicinata, è necessario per prevenire e trattare prontamente le complicanze della malattia, che presentano un’evoluzione lenta ed insidiosa specie per le funzioni muscolari e scheletriche. Nessun paziente riesce ad essere completamente autonomo e l’attesa di vita è ridotta.
Terapia Enzimatico Sostitutiva
Fino a poco tempo fa l’unica opzione terapeutica “specifica” per la alfa-Mannosidosi era il trapianto del midollo osseo (6), anche se le esperienze riportate sono limitate, sia nel numero dei pazienti trattati che dei risultati clinici, condizionati dall’entità del coinvolgimento neurologico e dell’apparato scheletrico.
Recentemente è stata messa a punto una terapia enzimatico sostitutiva (Enzyme Replacement Therapy, ERT) anche per la alfa-Mannosidosi (7, 8, 9, 10). Nel gennaio 2018 la European Medicines Agency ha approvato la commercializzazione nell’Unione europea del farmaco velmanase alfa, un enzima ricombinante per la terapia dei pazienti adulti, adolescenti e bambini affetti dalle forme lievi-moderate di alfa-Mannosidosi (Fig.2).

La limitazione è condizionata dell’impossibilità dell’enzima ricombinante di attraversare la barriera emato-encefalica (BBB), similmente a quanto già evidenziato per altre patologie lisosomiali.
Nella ERT per la alfa-Mannosidosi comunque era stata osservata, negli studi preclinici su modello murino, una significativa riduzione degli oligosaccaridi accumulati nel cervello. Uno studio successivo effettuato sui topi knockout per la alfa- mannosidasi, trattati a dosaggi elevati di ERT, dimostrava che l’enzima era in grado di passare la BBB e di entrare nei neuroni. Una possibile spiegazione dell’osservazione potrebbe risiedere nell’attivazione di un meccanismo di endocitosi a dosaggi molto alti di ERT, tale da permettere il passaggio della BBB (11, 12).
Nel 2013 lo studio di Borgwardt L et al. di fase I-II, che includeva 10 pazienti di età compresa fra 7 e dieci anni trattati per 12 mesi con velmanase alfa, ha dimostrato la sicurezza e l’efficacia della terapia a due diversi dosaggi: 25 e 50 U/Kg (7). L’enzima è stato ben tollerato senza reazioni secondarie significative e ha dimostrato clinicamente il miglioramento delle performances motorie nei pazienti trattati, misurate mediante il 3 Minute Stair Climbing test (3-MSCT), e la progressiva riduzione del livello degli oligosaccaridi liquorali nei 12 mesi di trattamento. Non sono stati riscontrati miglioramenti nella funzione uditiva. Questi risultati sono stati successivamente confermati da uno studio in fase III effettuato su 14 pazienti adulti (8).
Più recentemente gli studi di Lund AM et al. (10) e Harmatz P et al. (13) hanno analizzato in maniera comparativa i risultati degli studi registrativi (fasi I/ II e III) di velmanase alfa su 33 pazienti (19 pediatrici e 14 adulti) con un follow-up di lunga durata, fino a 4 anni (media 29 mesi). Gli end-point valutati riguardavano sempre le funzioni motorie e la riduzione biochimica degli oligosaccaridi sierici. I risultati hanno confermato, anche a lungo tempo, il significativo miglioramento dei test motori (3-MSCT e 6-minute walking test, 6-MWT) e della funzionalità respiratoria (FVC) in particolare nei pazienti in età pediatrica, mentre i pazienti adulti mostravano una stabilizzazione funzionale fino a 2 anni dall’inizio del trattamento ed una significativa riduzione degli oligosaccaridi nel siero.
Questi dati confermano le osservazioni già evidenziate per altre malattie lisosomiali di una differenza di risposta terapeutica legata alla precocità dell’intervento e all’età dei pazienti, sottolineando ulteriormente la positività di una diagnosi precoce, da sviluppare anche nel contesto futuro di uno screening neonatale per le malattie lisosomiali.
Bibliografia
- Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis. 2008; 3: 21.
- Meikle Pj et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004; Oct; 114(4): 909-16.
- Platt FM. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat Rev Drug Discov. 2018;17:133–150.
- Ceccarini MR, Codini M, Conte C, Patria F, Cataldi S, Bertelli M, Albi E, Beccari T. Alpha-Mannosidosis: Therapeutic Strategies. Int J Mol Sci. 2018, 19: pii: E1500.
- Riise Stensland HM, Frantzen G, Kuokkanen E, Buvang EK, Klenow HB, Heikinheimo P, Malm D, Nilssen Ø. amamutdb.no: A relational database for MAN2B1 allelic variants that compiles genotypes, clinical phenotypes, and biochemical and structural data of mutant MAN2B1 in -mannosidosis. Hum Mutat. 2015; 36: 581-586.
- Wall DA, Grange DK, Goulding P et al. Bone marrow transplantation for the treatment of a-mannosidosis. J Pediatr 1998; 133: 282–285.
- Borgwardt L, Dali CI, Fogh J, Månsson JE, Olsen KJ, Beck HC, Nielsen KG, Nielsen LH, Olsen SO, Riise Stensland HM, et al. Enzyme replacement therapy for alpha-mannosidosis: 12 months follow-up of a single centre, randomised, multiple dose study. J Inherit Metab Dis 2013; 36: 1015-1024.
- Borgwardt L, Lund AM, Amraoui Y, Andersen O, De Meirleir L, Dolhem P, Campos MG, Guffon N, Héron B, Laroche C, et al. Long-term enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase) slows disease progression in adult patients suffering from alpha-mannosidosis. Mol Genet Metab. 2017; 120: s30.
- Borgwardt L, Guffon N, Amraoui Y, Dali CI, De Meirleir L, Gil-Campos M, Heron B, Geraci S, Ardigò D, Cattaneo F, et al. Efficacy and safety of velmanase alfa in the treatment of patients with alfa-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind randomized, placebo-controlled trial. J Inherit Metab Dis. 2018; 41: 1215-1223.
- Lund AM, Borgwardt L, Cattaneo F, Ardigò D, Geraci S, Gil-Campos M, De Meirleir L, Laroche C, Dolhem P, Cole D, et al. Compreensive long-term efficacy and safety of recombinant human alpha-mannosidase (velmanase alfa) treatment in patients with alpha-mannosidosis. J Inherit Metab Dis. 2018; 41: 1225-1233.
- Damme M et al. Chronic enzyme replacement therapy ameliorates neuropathology in alpha-mannosidosis mice. Ann Clin Transl Neurol. 2015; 2: 987–1001
- Stroobants S et al., Long-term enzyme replacement therapy improves neurocognitive functioning and hippocampal synaptic plasticity in immune-tolerant alpha-mannosidosis mice. Neurobiol Dis. 2017;106:255-268. doi: 10.1016/j.nbd.2017.07.013. Epub 2017 Jul 15.
- Harmatz P et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol. Gen Met. 2018; 124: 152-160.