




la Newsletter

Andrea Guala1, Alexandra Liava2, Angelina Cistaro3, Maria Elena Liverani4, Michela Malacarne5, Chiara Baldo5, Domenico Coviello5, Cesare Danesino6
1SOC Pediatria, Ospedale Castelli, Verbania; 2SOC NPI, Ospedale Castelli, Verbania; 3Servizio di Medicina Nucleare, Salus Alliace Medical, Genova; 4già UOC Pediatria, Azienda Ospedaliera Universitaria S. Andrea, Roma; 5Laboratorio di Genetica Umana, IRCSS Giannina Gaslini, Genova; 6Dipartimento di Medicina Molecolare, Università di Pavia, Pavia
Novità sulla sindrome del Cri du Chat: un modello di studio per...
Le caratteristiche cliniche della sindrome del Cri du Chat...
La sindrome del Cri du Chat (CdC) (OMIM 123450), descritta per la prima volta da Lejeune e collaboratori nel 1963, è associata ad una delezione del braccio corto del cromosoma 5. L’incidenza va da 1:15.000 a 1:50.000 nati vivi; attualmente in Italia si segnalano 2-4 casi all’anno. Le caratteristiche cliniche tipiche includono un pianto acuto, simile al miagolio di un gatto, microcefalia, un fenotipo facciale che si modifica con l’età ed un severo ritardo psicomotorio e mentale, compromesso su vari versanti, che correla con le dimensioni della delezione sul cromosoma 5, con delezioni e/o duplicazioni in altri cromosomi (presenti in circa il 10-20 % dei casi), e con fattori ambientali quali una presa in carico educativa precoce, soprattutto se intensiva ed a domicilio.
Negli ultimi 20 anni sono state acquisite nuove informazioni sulla malattia che hanno modificato la presa in carico e la storia clinica.
Genetica
La comparsa della malattia non correla con l'età materna o paterna ma la maggior parte delle delezioni coinvolge il cromosoma paterno; nell’80-90% dei casi è terminale, nel 10-20% è interstiziale. Circa l’80% delle delezioni è sporadica, il 10% deriva da una traslocazione parentale bilanciata, il 10% è causato da riarrangiamenti cromosomici più rari. Si è individuata una regione critica tra le bande 5p15.2 e 5p15.3; la banda 5p15.2 sembra associata a microcefalia, deficit intellettivo e alcune caratteristiche facciali, la banda 5p15.3 al caratteristico pianto ed al ritardo del linguaggio. Ulteriori studi hanno individuato geni candidati per specifici segni clinici: CTNND2 è stato associato a deficit intellettivo grave, SEMA5A sembrerebbe associato ai disturbi dello spettro autistico, TERT alla variabilità fenotipica.
La diagnosi genetica può essere fatta mediante cariotipo o, meglio, mediante Array-CGH.
Dai primi anni ‘90 l’Associazione Bambini CdC ha supportato la creazione di una raccolta di campioni biologici di quasi 150 pazienti, e in molti casi anche dei genitori, oggi gestita dalla Biobanca del Laboratorio di Genetica Umana dell’IRCCS Gaslini di Genova.
Aspetti neurologici e neuroimaging
 Le tappe dello sviluppo psicomotorio sono rallentate, ma la maggior parte dei giovani adulti è in grado di comunicare e di deambulare autonomamente. Tutti i pazienti presentano segni cerebellari, disprassia globale e del distretto oro-linguale e rinolalia; la microcefalia è costante. Il ritardo cognitivo è più spesso medio o grave; ma le performances psicosociali dipendono molto dalla precocità ed intensità della presa in carico psicomotoria ed educativa (1). A volte i ragazzi sono autonomi nell'igiene personale e nell'alimentazione; molti sono in grado di formulare frasi complete, alcuni hanno una produzione linguistica limitata a singole parole o suoni finalizzati. Uno studio italiano (submitted, 2021) sulla produzione vocale in 63 bambini ed adulti con CdC (15 bambini maschi, 13 bambine femmine, 20 maschi adulti, 15 femmine adulte) ha dimostrato una serie di anomalie della produzione della voce che possono guidare la riabilitazione logopedica migliorando l'intelligibilità comunicativa (Tab. 1).
Le tappe dello sviluppo psicomotorio sono rallentate, ma la maggior parte dei giovani adulti è in grado di comunicare e di deambulare autonomamente. Tutti i pazienti presentano segni cerebellari, disprassia globale e del distretto oro-linguale e rinolalia; la microcefalia è costante. Il ritardo cognitivo è più spesso medio o grave; ma le performances psicosociali dipendono molto dalla precocità ed intensità della presa in carico psicomotoria ed educativa (1). A volte i ragazzi sono autonomi nell'igiene personale e nell'alimentazione; molti sono in grado di formulare frasi complete, alcuni hanno una produzione linguistica limitata a singole parole o suoni finalizzati. Uno studio italiano (submitted, 2021) sulla produzione vocale in 63 bambini ed adulti con CdC (15 bambini maschi, 13 bambine femmine, 20 maschi adulti, 15 femmine adulte) ha dimostrato una serie di anomalie della produzione della voce che possono guidare la riabilitazione logopedica migliorando l'intelligibilità comunicativa (Tab. 1).
Ai fini di specificare il fenotipo neuroradiologico e correlarlo a specifiche delezioni, sono state valutate le RMN di 14 pazienti (2). Il reperto più comune è l’ipoplasia isolata del ponte (13/14), seguita dall’ipoplasia del corpo calloso (11/14) e del verme cerebellare (7/14), da anomalie dei ventricoli laterali (10/14) e da alterazioni di struttura della corteccia cerebrale (6/14), per lo più polimicrogiria frontoparietale bilaterale (4/6). Altro reperto, mai riportato, è stata l’ipoplasia del nervo ottico (3/14). In questo studio, infine, si è evidenziato che la regione 5p15.33-15.2 è associata all’ipoplasia del ponte, mentre altre regioni cromosomiche sono correlate all’ipoplasia del verme ed alle anomalie dei ventricoli e dello sviluppo corticale.
Spesso i genitori riportano una ridotta sensibilità dolorifica al calore. Questo riscontro correla con lesioni cutanee autoinflitte anche gravi soprattutto sulle braccia e sulle mani (skin picking, SP) (3). Lo SP è presente nell’85% dei casi e può esordire già nel primo anno di vita. Le anomalie cerebellari e le alterate vie di comunicazione fra cervelletto e corteccia sono una possibile causa dello SP.
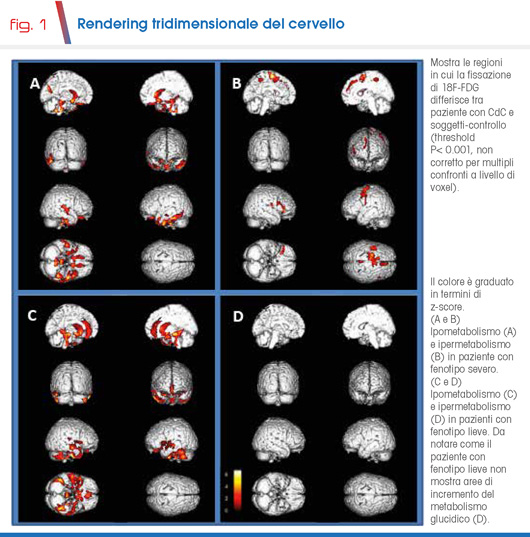 Un recente lavoro su 6 soggetti ha valutato il metabolismo cerebrale mediante la tomografia ad emissioni di positroni (PET). Lo studio ha mostrato una significativa riduzione del metabolismo, in parte correlabile con alcune caratteristiche cognitivo-comportamentali, nel lobo temporale di sinistra; è stato anche evidenziato un incremento del metabolismo nella corteccia motoria destra dei soggetti con fenotipo severo (Fig. 1) (4).
Un recente lavoro su 6 soggetti ha valutato il metabolismo cerebrale mediante la tomografia ad emissioni di positroni (PET). Lo studio ha mostrato una significativa riduzione del metabolismo, in parte correlabile con alcune caratteristiche cognitivo-comportamentali, nel lobo temporale di sinistra; è stato anche evidenziato un incremento del metabolismo nella corteccia motoria destra dei soggetti con fenotipo severo (Fig. 1) (4).
Risultati analoghi sono stati segnalati in due gemelli monozigoti, mosaici per la delezione 5p, in cui un incremento del metabolismo corticale è presente solo nel fratello con fenotipo severo (submitted, 2021). Sembrerebbe che l’ipermetabolismo corticale rifletta una neuroinfiammazione associata ad attivazione astrocitaria o microgliale ed abbia un ruolo nell'espressione delle caratteristiche cliniche più gravi legate al movimento.
Follow-up clinico
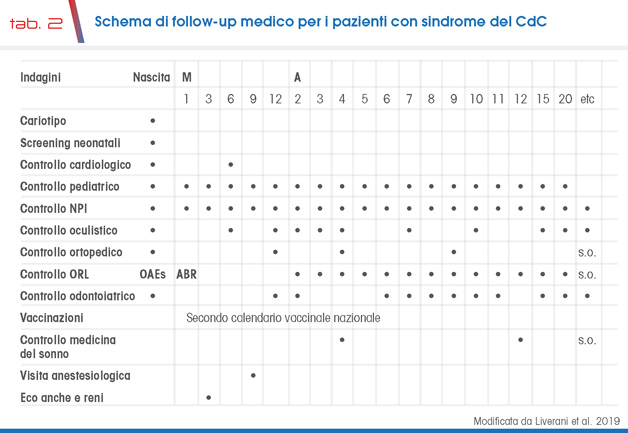
Alcuni problemi sono più frequenti nei pazienti con sindrome CdC; occorre quindi che i pazienti siano seguiti da specialisti che conoscono la sindrome, la sua evoluzione ed i limiti della “care”. La tabella 2 schematizza il follow-up utile per anticipare la diagnosi delle principali problematiche il più precocemente possibile alle diverse età.
La scoliosi è frequente ed ha un’evoluzione rapida, soprattutto nell’adolescenza. Occorre un controllo serrato per una diagnosi precoce e una presa in carico da parte di un ortopedico che conosca la sindrome.
Il fenotipo occlusale è spesso anomalo (affollamento, morso aperto, palato ogivale) e può condizionare sia la funzione deglutitoria che la respirazione notturna, compromettendo la qualità del sonno; inoltre le malocclusioni e la scarsa igiene orale incrementano il rischio di carie, che provoca una sintomatologia dolorosa a volte non precisamente localizzata ed episodi infettivi. Occorre quindi una precoce presa in carico odontoiatrica presso un centro competente nella gestione dei disabili.
Un buon visus consente di sfruttare al meglio le opportunità dei nuovi approcci riabilitiativo-pedagogici (quali la comunicazione aumentativa alternata o la comunicazione facilitata) e di apprezzare un programma televisivo, sfogliare un fumetto, o giocare ad un videogioco. Cataratta, grave miopia, problemi retinici e del nervo ottico possono compromettere la funzione visiva dei pazienti CdC. Non appena la collaborazione del bambino lo consenta è consigliato un follow-up oculistico periodico.
Sono stati descritti deficit sensoriali che dipendono da un alterato funzionamento del SNC e meritano un corretto inquadramento diagnostico. Un SNC “travolto” da informazioni che non riesce ad organizzare non accetta insegnamenti né può discriminare stimoli “importanti per la sopravvivenza”.
Un aspetto peculiare è quello anestesiologico. Il tipico pianto neonatale è stato considerato indice di anomalie anatomiche della laringe, delle corde vocali e dell’epiglottide allarmando gli anestesisti per le procedure di intubazione. Una recente revisione su 51 pazienti sottoposti ad 80 anestesie non ha però riportato particolari problemi (5).
Dato che fino al 40% dei pazienti potrebbe necessitare prima o poi di sedazione, è consigliabile una visita anestesiologica presso l’ospedale di riferimento più vicino al domicilio all’interno dei bilanci di salute. Inoltre il paziente dovrà portare con sé una “anestetic card”, redatta dagli anestesisti che conoscono il paziente e che riporti i principali problemi riscontrati.
La gestione del paziente in età adulta
La mortalità, del 10% circa, si concentra nel primo anno di vita; superato questo periodo la sopravvivenza è prolungata.
Il raggiungimento dell'età adulta ed anziana espone i pazienti alle patologie che affliggono i soggetti di pari età. Un’indagine su 321 casi censiti dalle associazioni di pazienti italiana e tedesca non ha identificato predisposizioni a particolari patologie neoplastiche (6).
I genitori di pazienti adulti segnalano che le competenze acquisite tendono a perdersi, verosimilmente per la progressiva diminuzione degli stimoli con il passaggio dalla scuola ai centri diurni. Sembra che i ragazzi richiedano “allenamento costante”, che non può essere esclusivamente delegato ai genitori.
Occorrono quindi percorsi riabilitativi ed educativi specifici per adulti ed anziani. Purtroppo ci si trincera frequentemente dietro ad una supposta scarsa collaborazione del paziente per limitarsi a prestazioni scadenti. L’Associazione Bambini CdC offre un progetto educativo per formare le famiglie e gli operatori e ha registrato 25 conferenze su questi argomenti disponibili sul proprio canale YouTube.
Pedagogisti e terapisti dell’associazione seguono le famiglie e coordinano il programma riabilitativo collaborando con le scuole ed i servizi socio-assistenziali.
La crescita in famiglia, il precoce inizio delle stimolazioni, la fisioterapia, l’uso di tecnologie informatiche e la pratica sportiva hanno migliorato la qualità di vita e l’inserimento sociale dei pazienti e delle famiglie come dimostrato da un'indagine che ha indagato il benessere, la disabilità ed i costi diretti ed indiretti in 76 gruppi di pazienti/famiglie italiani (7).
Infine occorre citare gli abusi (sessuali, psicologici, fisici, da incuria o eccesso di cure) di cui possono essere oggetto i pazienti CdC che spesso non sono in grado di riconoscere e denunciare situazioni di sofferenza. A volte capita anche che il paziente abusi della pazienza e disponibilità dei caregivers rendendo insopportabile il loro carico assistenziale. È necessario valutare anche questi aspetti individuando i segnali di pericolo nei comportamenti quotidiani e ascoltando le richieste di aiuto della famiglia.
Per maggiori informazioni: Associazione Bambini CdC, www.criduchat.it, tel. 055-828683, mail: abc@criduchat.it.
Bibliografia
- Guala A, Spunton M, Tognon F, et al. Psychomotor development in Cri du Chat syndrome: comparison in two Italian cohorts with different rehabilitation methods. Sci World J. 2016;16:3125283.
- Villa R, Fergnani VGC, Silipigni R, et al. Structural brain anomalies in Cri-du-Chat syndrome: MRI findings in 14 patients and possible genotype-phenotype correlations. Eur J Paediatr Neurol. 2020;28:110-119.
- Spunton M, Guala A, Liverani ME, et al. Skin picking disorder in 97 Italian and Spanish Cri du chat patients. Am J Med Genet A. 2019;179:1525-1530.
- Cistaro A, Quartuccio N, Piccardo A, et al. 18F-fluorodexyglucose Position Emission Tomography identifies altered brain metabolism in patients with Cri du Chat syndrome. J Nucl Med. 2020;61:1195-9.
- Guala A, Spunton M, Cerruti Mainardi P, et al. Anesthesia in Cri du Chat syndrome: Information on 51 Italian patients. Am J Med Genet. 2015;167A:1168-70.
- Guala A, Spunton M, Kalantari S, et al. Neoplasia in Cri du Chat Syndrome from Italian and German Databases. Case Rep Genet. 2017;17:5181624.
- Kodra Y, Cavazza M, de Santis M, Guala A, et al. Social Economic Costs, Health-Related Quality of Life and Disability in Patients with Cri Du Chat Syndrome. Int J Environ Res Public Health. 2020;17:5951.