




la Newsletter
Quando un WES può indirizzare la clinica: un paziente con...
La sindrome di Rubinstein-Taybi è caratterizzata da tratti...
Conosciamo C. all’età di 6 mesi nel corso di una visita in Ambulatorio di Genetica Clinica Pediatrica, inviato dallo specialista chirurgo maxillo-facciale che lo aveva valutato per labio-palatoschisi (LPS)
C. è unicogenito di genitori sani non consanguinei. Una precedente gravidanza è esitata in MEF; l’autopsia non aveva evidenziato malformazioni maggiori. L’anamnesi familiare è negativa per malattie genetiche o quadri malformativi. Nello specifico non sono riportati altri soggetti con labio-palatoschisi nella famiglia. La malformazione è stata evidenziata nel corso della gravidanza, durante i controlli ecografici, cariotipo ed arrayCGH non hanno evidenziato alterazioni patologiche.
Nato da taglio cesareo per presentazione podalica alla 37°+3 s.g. , segnalato polidramnios. IUGR, il peso alla nascita era 2,350 Kg (3-10° P), lunghezza 43 cm (< 3° P), CC 31 cm (<3° P), l’indice di APGAR 9/10. Nei primi giorni di vita è stato sottoposto a ventilazione ad alti flussi per comparsa di distress respiratorio a poche ore dalla nascita.
E’ stata evidenziata una pervietà del dotto arterioso, con pervietà del forame ovale e arco aortico destro, con iniziale ipertensione polmonare. Fin dai primi giorni di vita, C. ha presentato iponatriemia persistente, tale da richiedere supplementazione con NaCl. Nella norma gli esami ormonali basali (ADH, aldosterone, cortisolo, ACTH, TSH), l’elettrolituria su 24 ore, il test del sudore, l’ecografia addominale. La RMN encefalo evidenziava solo una modesta riduzione della mielinizzazione senza alterazioni dello studio spettroscopico. Per difficoltà ingravescenti nella suzione con insufficiente incremento ponderale, all’età di 3 mesi è stata posizionata gastrostomia, con successiva ripresa della crescita.
 Al momento della nostra valutazione ambulatoriale, C. pesa 4,650 kg (<3° P), la lunghezza è di 55,5 cm (<3° P), la circonferenza cranica di 35 cm (<<3° P). Presenta una brachicefalia con plagiocefalia, ipertelorismo e fessure palpebrali lunghe, importante labio-palatoschisi bilaterale (Fig. 1).
Al momento della nostra valutazione ambulatoriale, C. pesa 4,650 kg (<3° P), la lunghezza è di 55,5 cm (<3° P), la circonferenza cranica di 35 cm (<<3° P). Presenta una brachicefalia con plagiocefalia, ipertelorismo e fessure palpebrali lunghe, importante labio-palatoschisi bilaterale (Fig. 1).
Si evidenziano un discreto ipotono assiale ed edemi a carico del dorso di mani e dei piedi. L’obiettività toracica e addominale è nella norma. Al cuore: presenta impurità sistolica mentre a livello dei genitali è evidente micropene, criptorchidismo bilaterale.
Il piccolo è stato ricoverato per inquadramento diagnostico presso la nostra struttura. E’ stata ripetuta RMN encefalo che ha documentato parziale agenesia del corpo calloso (ginocchio e rostro), ventricoli laterali ampliati, parziale fusione dei fornici del setto pellucido, sospetta ipoplasia del tratto ottico di sinistra, sospetta agenesia del bulbo olfattorio sinistro, adenoipofisi e peduncolo normali ma iperintensità neuroipofisaria non chiaramente riconoscibile. La visita oculistica ha confermato sospetto deficit visivo corticale, il test ABR ha evidenziato lieve ipoacusia trasmissiva a sinistra. EEG nella norma. All’Rx dei piedi riscontro di assenza della falange intermedia del V dito bilateralmente.
Sono stati ripetuti esami per lo studio dell’asse ipotalamo-ipofisario, che hanno documentato normale funzione tiroidea e surrenalica; i valori di gonadotropine dopo test di stimolo sono risultati suggestivi per possibile ipogonadismo ipogonadotropo. IGF-1 inferiori alla norma, mai episodi di ipoglicemia. Gli accertamenti eseguiti hanno escluso un ipocorticismo e una “central salt wasting syndrome” quali cause della iponatriemia mentre i dati clinici e laboratoristici sembrano, invece, orientare per un quadro SIADH-like che ha risposto alla restrizione di liquidi. Il quadro è stato ben controllato mediante integrazione con NaCl.
All’età di 8 mesi è stato effettuato il primo intervento di correzione per labiopalatoschisi. Durante l’intervento si è osservata importante ipercapnia transitoria che è stata adeguatamente corretta.
Poiché il quadro clinico, pur essendo orientativo per una patologia genetica sindromica su base costituzionale, non permetteva di porre ipotesi diagnostiche specifiche è stato scelto di eseguire l’analisi dell’intero esoma sul probando ad entrambi i genitori (trio-WES). L’analisi è stata eseguita mediante ibridazione di sonde complementari alle regioni codificanti per proteine del genoma umano (esoma) al DNA del probando e dei genitori. Dopo il sequenziamento e l’elaborazione del dato grezzo, prodotto dallo strumento, mediante strumenti bioinformatici ad hoc, la lista delle varianti ottenute è stata filtrata. Questo passaggio consente di selezionare le varianti potenzialmente significative, considerando le caratteristiche cliniche del probando e il modello di trasmissione ipotizzato per la condizione nella famiglia. In assenza di familiarità, il primo modello che si testa è quello per le condizioni sporadiche, causate solitamente da mutazioni de novo (assenti nei genitori) nell’esoma del probando.
Nei mesi successivi, C. ha presentato respiro rumoroso con necessità di frequenti aspirazioni delle prime vie respiratorie e frequenti episodi di infezioni delle alte vie, trattate con antibioticoterapia a domicilio; è stato nuovamente ricoverato per infezione delle vie urinarie da E. coli multiresistente, la cistoscintigrafia è risultata nella norma.
Il piccolo ha effettuato controlli seriati dell’equilibrio acido-base e degli elettroliti, che hanno evidenziato progressiva comparsa di alcalosi metabolica e ipocloremia con iponatriemia, e incremento di renina e aldosterone; parziale risposta alla restrizione di liquidi ma graduale ripresa della normalità dopo somministrazione di NaCl, anche per quanto riguarda renina e aldosterone.
Il risultato dell’analisi WES è giunto alla nostra attenzione quando C. aveva 16 mesi. L’indagine ha mostrato la presenza della variante de novo p.Arg483ter nel gene CREBBP. Tale variante porta alla produzione di una proteina tronca e ha suggerito la diagnosi di sindrome di Rubinstein-Taybi (RTS).
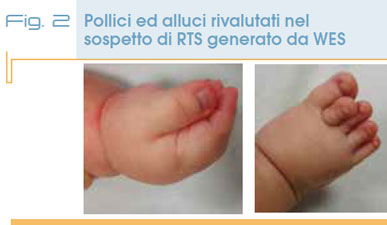 L’esito dell’indagine genomica ha comportato un’attenta rivalutazione del fenotipo del bambino, grazie a cui abbiamo valorizzato uno slargamento di pollici e alluci (Fig. 2), caratteristiche tipiche della RTS che, associate alle già note anomalie dell’accrescimento, dello sviluppo psicomotorio, alle anomalie genitali, labio-palatine e cardiache, supportavano il sospetto di RTS.
L’esito dell’indagine genomica ha comportato un’attenta rivalutazione del fenotipo del bambino, grazie a cui abbiamo valorizzato uno slargamento di pollici e alluci (Fig. 2), caratteristiche tipiche della RTS che, associate alle già note anomalie dell’accrescimento, dello sviluppo psicomotorio, alle anomalie genitali, labio-palatine e cardiache, supportavano il sospetto di RTS.
Una volta avvenuta la correzione chirurgica della malformazione facciale le stesse caratteristiche del volto del bambino, grazie anche all’evoluzione nei mesi della sua fisionomia sono risultate più convincenti per la diagnosi suggerita dall’indagine genomica di RTS.
La sindrome di Rubinstein-Taybi (RTS)
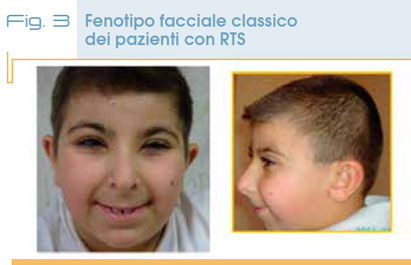 Ha un’incidenza di 1:100.000 – 125.000 nati vivi, è una sindrome genetica caratterizzata da tratti somatici peculiari, alluci e/o pollici slargati e angolati, bassa statura e ritardo di sviluppo di grado moderato o severo. Le anomalie minori che costituiscono la gestalt facciale comprendono fessure palpebrali orientate verso il basso dall’ esterno all’interno, radice nasale alta con columella prominente al di sotto delle ali del naso, palato ogivale, cuspidi appuntite sul lato interno degli incisivi permanenti. L’espressione del viso delle persone con RTS è assai specifica; è soprattutto caratteristico il sorriso con chiusura quasi completa delle rime palpebrali (Fig. 3).
Ha un’incidenza di 1:100.000 – 125.000 nati vivi, è una sindrome genetica caratterizzata da tratti somatici peculiari, alluci e/o pollici slargati e angolati, bassa statura e ritardo di sviluppo di grado moderato o severo. Le anomalie minori che costituiscono la gestalt facciale comprendono fessure palpebrali orientate verso il basso dall’ esterno all’interno, radice nasale alta con columella prominente al di sotto delle ali del naso, palato ogivale, cuspidi appuntite sul lato interno degli incisivi permanenti. L’espressione del viso delle persone con RTS è assai specifica; è soprattutto caratteristico il sorriso con chiusura quasi completa delle rime palpebrali (Fig. 3).
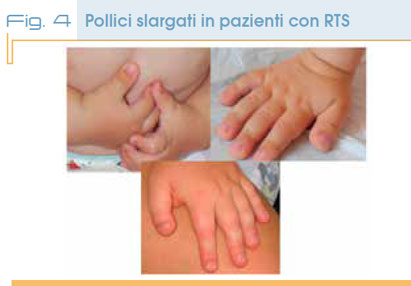 A livello delle estremità oltre alla tipica forma slargata o bifida degli alluci e dei pollici (in un terzo dei casi associata a deviazione radiale (Fig. 4), anche le falangi terminali delle altre dita possono essere slargate.
A livello delle estremità oltre alla tipica forma slargata o bifida degli alluci e dei pollici (in un terzo dei casi associata a deviazione radiale (Fig. 4), anche le falangi terminali delle altre dita possono essere slargate.
Si possono associare inoltre anomalie maggiori a carico di vari organi ed apparati:
- Anomalie cardiache (24-48%): nella maggioranza come difetto unico (pervietà del dotto arterioso, difetti settali, coartazione o stenosi della polmonare).
- Anomalie del Sistema Nervoso Centrale (5-25%): mielopatia per stenosi della giunzione cranio-vertebrale da alterazioni delle vertebre cervicali, malformazioni di Chiari, siringomielia, midollo ancorato. Nel 74% dei casi: alterazioni del corpo calloso.
- Anomalie dell’apparato genitourinario e renale (50-75%): criptorchidismo, ipospadia, alterazioni renali causa di frequenti infezioni delle vie urinarie.
In termini auxologici la crescita prenatale generalmente è nella norma, nel periodo post-natale l’altezza e la circonferenza cranica sono solitamente al di sotto dei limiti di norma tanto che la presenza di sovrappeso/franca obesità è una delle complicanze più frequenti di questa condizione e tende a manifestarsi durante l’infanzia nei maschi e durante l’adolescenza nelle femmine.
Lo sviluppo puberale e sessuale è normale. A livello ormonale sono descritti singoli pazienti con deficit di GH, ipotiroidismo congenito, ipoplasia ipofisaria.
Lo sviluppo psicomotorio è caratterizzato da un ritardo globale che generalmente varia da una gravità moderata a severa; è comunque presente una discreta variabilità individuale. Nei giovani-adulti insorgono frequentemente disturbi dell’umore, ansia e alterazioni comportamentali.
In termini di complicanze mediche non raro è il riscontro di problematiche ortopediche quali dislocazione della rotula, iperlassità legamentosa, anomalie di curvatura della colonna, petto escavato, morbo di Perthes. È presente epilessia nel 25-27% dei casi, ma le anomalie all’EEG sono riscontrabili in un terzo dei casi. Dal punto di vista ORL sono discretamente frequenti le apnee ostruttive del sonno e le problematiche a carico dell’orecchio medio (50%), con ipoacusia transmissiva che spesso peggiora una già coesistente ipoacusia neurosensoriale (25%). A livello oculare possono essere presenti strabismo (33-58%), difetti di rifrazione (41%), ptosi palpebrale, ostruzione dei dotti lacrimali, cataratta, coloboma, nistagmo, glaucoma e anomalie corneali. Con l’aumentare dell’età insorgono disfunzioni retiniche nel 78% dei pazienti, dimostrabili con elettroretinogramma. A livello cutaneo possibile formazione di cheloidi in seguito a traumi cutanei anche minimi. Occasionale ma non raro l’insorgenza di pilomatrixomi. I pazienti con RTS mostrano anche svariate possibili anomalie odontoiatriche quali sovraffollamento dentale, malocclusione, carie multiple, ipodontia, iperdontia, denti neonatali e le cuspidi caratteristiche a livello degli incisivi superiori. In letteratura sono riportati pazienti con sindrome di Rubinstein Taybi e labio-palatoschisi in associazione ad alterazione del gene CREBBP.
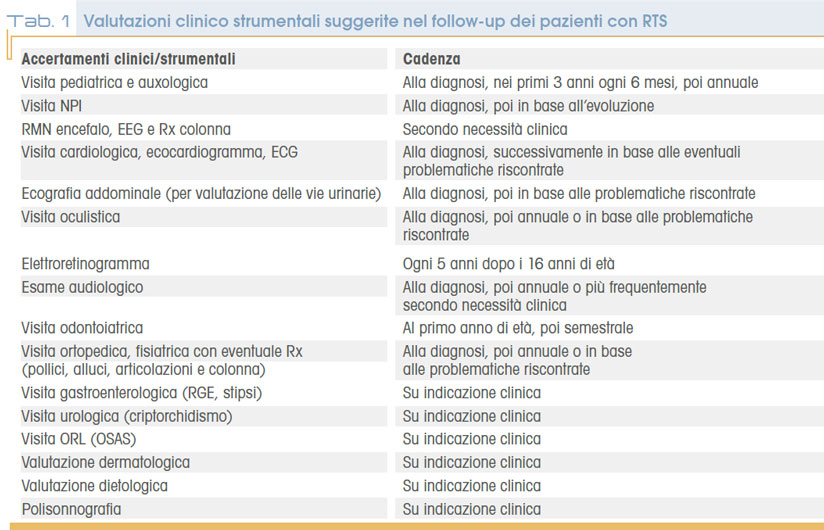
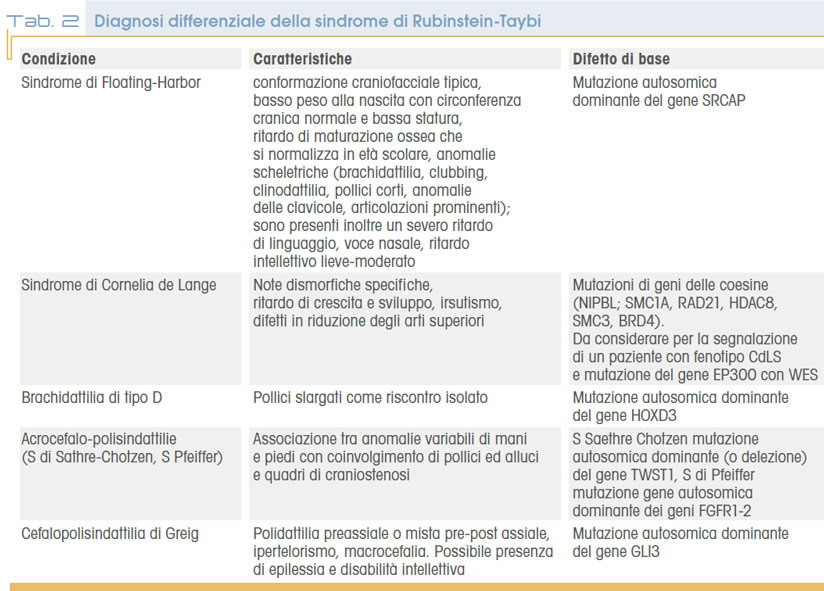
Molto frequentemente (80%) i lattanti con RTS manifestano difficoltà nella suzione e deglutizione, a volte con necessità di alimentazione enterale (tramite sondino naso gastrico o PEG). Talora si associa reflusso gastro-esofageo (25-50%), spesso con sintomatologia atipica come agitazione notturna, scialorrea, tosse stizzosa o infezioni respiratorie ricorrenti. E’ stata infine dimostrata una correlazione fra RTS e incrementata incidenza di neoplasie benigne e maligne di varia origine (più frequentemente derivanti dalla cresta neurale), quali meningiomi e altri tumori encefalici, pilomatrixomi, rabdomiosarcomi narofaringei, neurilemmomi intraspinali, feocromocitomi, seminomi, leucemie e carcinomi mammari. Spesso tali tumori si manifestano nell’età dell’infanzia, prima dei 15 anni; più frequenti nell’età adulta i meningiomi. La predisposizione tumorale viene ricondotta al ruolo di CREBBP e EP300 (geni malattia della condizione) in alcuni pathways di soppressione tumorale. A differenza di altre sindromi “cancer predisposing”, nella RTS non è definito un protocollo di follow-up a causa della variabilità di sede dei tumori descritti. La tabella 1 mostra i principali accertamenti strumentali e clinici previsti nel follow-up della sindrome di Rubinstein-Taybi, la tabella 2 raccoglie le condizioni cliniche da considerare in diagnosi differenziale..
Eziologia
La sindrome di Rubinstein-Taybi è caratterizzata, come la gran parte delle sindromi genetiche più comuni, da una eterogeneità genetica. La condizione risulta infatti causata da microdelezioni a carico del braccio corto del cromosoma 16 (16p13.3) coinvolgenti il gene CREBBP (5-10%), mutazioni puntiformi di questo stesso gene (50-60%) e mutazioni del gene EP300 (5-8% circa). Nei soggetti con alterazioni del gene EP300, le caratteristiche peculiari del volto e delle estremità sono meno marcate e rendono più difficile il riconoscimento gestaltico; si associa solitamente un minore ritardo di sviluppo. L’utilizzo dei test citogenetico-molecolari combinati consente quindi di identificare il difetto di base in circa l’80% dei casi. Nella restante parte dei casi, soprattutto in caso di fenotipi lievi, bisogna tener presente l’eventualità di un mosaicismo somatico, che può essere dimostrato ripetendo le indagini molecolari su tessuti diversi dai linfociti del sangue periferico (analisi della saliva, del brushing buccale o dei fibroblasti cutanei). Il difetto genetico della condizione si trasmette con una modalità autosomica dominante; ciò significa che un soggetto affetto presenta un rischio del 50% di trasmettere il difetto genetico alla progenie indipendentemente dal sesso del nascituro. Nella maggioranza di casi la mutazione causativa insorge generalmente in modo casuale (de novo) nella linea germinale materna o paterna.
Da segnalare l’esistenza di un Associazione Italiana di genitori di soggetti con RTS: Associazione Italiana sindrome di Rubinstein-Taybi - Tel. 393/5817528 - www.rubinstein-taybi.it - e-mail: info@rubinstein-taybi.it.
Conclusioni
Nel paziente descritto l’analisi WES ha permesso di raggiungere la diagnosi di sindrome di Rubinstein Taybi in tempi rapidi in un soggetto in cui l’espressione del quadro dismorfologico era inizialmente fortemente condizionata dalla gravissima labio-palatoschisi e l’andamento clinico pediatrico caratterizzato dalle anomalie degli elettroliti a tutt’oggi di difficile inquadramento in ambito fisio-patologico. Come ormai ampiamente segnalato da più parti nella letteratura internazionale, l’analisi WES deve essere fortemente considerata come esame diagnostico fondamentale in tutte quelle situazioni in cui il quadro clinico è fortemente orientativo per una condizione genetico costituzionale ed in cui non è possibile porre ipotesi diagnostiche gestaltiche. Questo approccio permette infatti con una notevole efficienza (variabile da casistica a casistica tra il 30 ed il 50%) sia di identificare condizioni ultra rare la cui diagnosi e la cui conferma molecolare sono spesso fuori dalla portata di un percorso clinico tradizionale, sia di individuare condizioni note in pazienti in cui, come nel caso descritto, il fenotipo clinico mostra una espressione particolarmente atipica che mina il suo facile riconoscimento gestaltico.
Bibliografia
- Ajmone PF, Avignone S, Milani D, et al. Rubinstein–Taybi syndrome: New neuroradiological and neuropsychiatric insights from a multidisciplinary approach. Am J Med Genet. 2018;1–10.
- Bartsch O, Kress W, Kempf O, et al. Inheritance and variable expression in Rubinstein-Taybi syndrome. Am J Med Genet. PartA 2010;152A:2254-61.
- Bentivegna A, Milani D, Gervasini C, et al. Rubinstein-Taybi Syndrome: spectrum of CREBBP mutations in Italian patients. BMC Med Genet. 2006;7:77.
- Boot MV, van Belzen MJ, de Jong D, et al. Benign and malignant tumors in Rubinstein–Taybi syndrome. Am J Med Genet. 2018;1–12.
- Demeer B, Andrieux J, Mathieu-Dramard M, et al. Duplication 16p13.3 and the CREBBP gene: confirmation of the phenotype. Eur J Med Genet 2013 Jan;56(1):26-31.
- Dallapiccola B, Bernardini L, Mingarelli R, et al. Expanding the phenotype of duplication of the Rubinstein–Taybi region on 16p13.3. Am J Med Genet 2009; A 149A:2867–2870.
- Fergelot P, Van Belzen M, Hennekam RC, et al. Phenotype and genotype in 52 patients with Rubinstein–Taybi syndrome caused by EP300 mutations. Am J Med Genet 2016; 170:3069-3082.
- Hennekam RC, Rubinstein–Taybi syndrome. Eur J Hum Genet. 2006;14, 981–985.
- Hennekam RC, Van Doorne JM. Oral aspects of Rubinstein-Taybi syndrome. Am J Med Genet Suppl. 1990;6:42-47
- Levenson D. Whole-exome sequencing strategy proposed as first-line test: WES for well-phenotyped infants leads to high diagnostic yield Am J Med Genet A. 2016 Jun;170(6):1387-8
- Selicorni A, Zampino G, Memo L, et al. Le sindromi malformative: una guida per il pediatra. Pacini Editore Srl, 2017
- Tuysuz B, van Bon BW, de Vries BB, et al. A microduplication of the Rubinstein-Taybi region on 16p13.3 in a girl with a bilateral complete cleft lip and palate and severe mental retardation. Clin Dysmorphol 2012 Oct;21:204-7.
- Valencia CA, Husami A, Holle J, et al.Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience Front Pediatr. 2015 Aug 3;3:67.
- Wiley S, Swayne S, Rubinstein JH, et al. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119A:101-10.
- Woods S, Robinson HB, Khalifa M, et al. Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap Cornelia de Lange syndrome. Am J Med Genet A 2014; 164A:251–258.