




la Newsletter
Sindrome di Gorham-Stout, patologia complessa e assai rara di tipo...
La sindrome di Gorham-Stout (SGS), conosciuta anche come malattia...
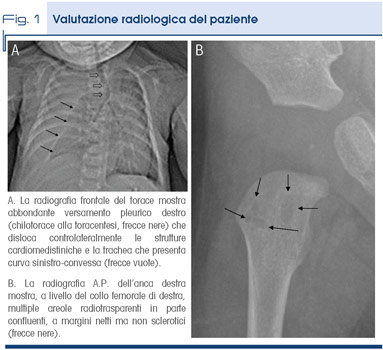 Un lattante di 3 mesi viene ricoverato in urgenza per la comparsa improvvisa di dispnea ingravescente. All'auscultazione toracica si apprezza ipofonesi estesa in emitorace destro e alla percussione ottusità nella stessa regione. La radiografia del torace (Fig. 1A) mostra un esteso versamento pleurico destro e, in via collaterale, permette di evidenziare lesioni osteolitiche multiple nella scapola destra. La radiografia dello scheletro completo, effettuata a completamento diagnostico, evidenzia analoghe lesioni a carico del sacro, di alcune vertebre (S1, L4, L5), dell'omero sinistro e del femore destro (Fig. 1B).
Un lattante di 3 mesi viene ricoverato in urgenza per la comparsa improvvisa di dispnea ingravescente. All'auscultazione toracica si apprezza ipofonesi estesa in emitorace destro e alla percussione ottusità nella stessa regione. La radiografia del torace (Fig. 1A) mostra un esteso versamento pleurico destro e, in via collaterale, permette di evidenziare lesioni osteolitiche multiple nella scapola destra. La radiografia dello scheletro completo, effettuata a completamento diagnostico, evidenzia analoghe lesioni a carico del sacro, di alcune vertebre (S1, L4, L5), dell'omero sinistro e del femore destro (Fig. 1B).
La valutazione iniziale include una toracentesi, che ha dimostrato fluido pleurico “bianco latte”. È stata quindi eseguita una biopsia della parte interessata dell'omero. L'esame istopatologico ha mostrato osteolisi con riscontro di cavità vascolari a parete sottile e tessuto connettivo fibroso ipervascolare. Sulla base dei risultati clinici, radiografici e istopatologici, viene diagnosticata la sindrome di Gorham-Stout.
Sindrome di Gorham-Stout
La sindrome di Gorham-Stout (SGS), conosciuta anche come malattia dell'osso fantasma, malattia dell'osso che scompare o osteolisi massiva idiopatica, è una malattia rara (circa 200 casi descritti al mondo). La rarità è probabilmente dovuta anche al fatto che spesso non viene correttamente diagnosticata.
La SGS è caratterizzata da un progressivo riassorbimento dell'osso dovuto a proliferazione vascolare o linfatica intraossea con conseguente scomparsa dell'osso che viene sostituito da tessuto fibroso.
Può interessare qualsiasi segmento osseo ma più frequentemente, nei casi descritti in letteratura, sono coinvolte le costole, le clavicole, le scapole, il bacino, il cranio e la colonna vertebrale.
L’età d’esordio descritta è anch’essa estremamente variabile: prevalentemente vengono descritti bambini e giovani adulti (< 40 anni), tuttavia sono riportati rari casi in età estreme (1 mese - 70 anni), senza predilezione di popolazione o sesso.
La sintomatologia d’esordio è quindi aspecifica, secondaria alla localizzazione: dolore osseo, debolezza muscolare, impotenza funzionale, tumefazione della regione affetta, deformità, fratture patologiche.
Quando, come nel caso descritto, la proliferazione interessa aree contigue alla pleura, può determinare versamento chiloso che compare come sintomo acuto. In caso di coinvolgimento vertebrale può comparire paralisi mentre l'interessamento delle ossa craniche può determinare l'insorgenza di meningiti.
Le complicanze più frequentemente descritte sono osteomielite e dolore cronico.
Diagnosi
La diagnosi di SGS è spesso di esclusione, in assenza di marker biochimici, istologici e genetici patognomonici e perciò può osservarsi una latenza anche di anni tra la comparsa dei sintomi e la diagnosi. In assenza di sintomi può essere addirittura occasionale in concomitanza con esami radiologici effettuati per altre cause.
I criteri per la diagnosi a cui ancora si fa riferimento sono effettivamente datati e non prevedono il contributo prezioso dei dati radiologici di nuova generazione, tuttavia sono utili a sottolineare i principali elementi patognomonici (1):
- presenza di tessuto angiomatoso
- assenza di atipia cellulare
- minima o assente risposta osteoclastica ed assenza di calcificazioni distrofiche
- evidenza di progressivo riassorbimento osseo localizzato a livello della lesione
- assenza di lesioni espansive o ulcerative
- assenza di coinvolgimento viscerale (salvo versamento chiloso)
- pattern osteolitico radiologico
- esclusione di cause metaboliche, neoplastiche, immunologiche ed infettive.
Gli esami di laboratorio, generalmente nella norma, possono essere di supporto per escludere altre cause di riassorbimento osseo.
La diagnosi si basa sull’insieme delle caratteristiche cliniche, radiologiche e sugli aspetti istopatologici.
La sindrome di Gorham-Stout è infatti difficilmente diagnosticabile tramite semplice valutazione clinica necessitando comunque di un completamento diagnostico tramite esame istologico e imaging (ecografia, CT e RM, scintigrafia, MOC) che permetteranno di acquisire tutte le informazioni per la definizione delle lesioni.
L'evoluzione clinica è imprevedibile: in alcuni casi il riassorbimento osseo rimane localizzato mentre in altri coinvolge progressivamente altri segmenti ossei anche non contigui. Le cause che determinano questa condizione clinica così polimorfa non sono chiaramente conosciute.
Inizialmente Gorham e Stout hanno ipotizzato che una lesione ipervascolare determinasse una massiva iperemia con successiva prevalenza di fenomeni di riassorbimento (attivazione degli osteoclasti) rispetto a quelli di produzione ossea (2). Più recentemente si è chiarito il contributo di vari fattori sistemici (interleuchine, fattori di crescita) in grado sia di modulare l’attività di osteoclasti e osteoblasti che la proliferazione vascolare (3-4).
Prospettive terapeutiche
Attualmente non è disponibile una terapia risolutiva. Le opzioni terapeutiche prevedono terapie mediche sintomatiche ed approcci di tipo chirurgico. Questi ultimi sono indicati quando si verificano fratture patologiche o nella ricostruzione ossea in caso di riassorbimento osseo massivo.
Anche l'irradiazione - in casi selezionati e soprattutto in età adulta - si è dimostrata efficace, in associazione o meno con la chirurgia, nel ridurre il dolore e nel rallentare la diffusione del riassorbimento osseo. Sono in corso tuttavia promettenti studi sperimentali che utilizzano farmaci che bloccano il riassorbimento osseo e la crescita incontrollata dei vasi linfatici e sanguigni, in particolare inibitori di mTor (repamicina e derivati) in associazione o meno con bifosfonati (5).
Bibliografia
- Heffez L, Doku HC, Carter BL, Feeney JE. Perspectives on massive osteolysis. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1983;55(4):331-43.
- Gorham LW, Stout AP Massive osteolysis: its relation to hemangiomatosis. J Bone Joint Surg Am. 1955; 37 (5): 985-1004.
- Hirayama T, Sabokbar A, Itonaga I, et al. Cellular and humoral mechanism of osteoclast formation and bone resorption in Gorham-Stout disease. J Pathol. 2001;195(5):624-30.
- Rossi M, Buonuomo PS, Battafarano G, et al. Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone. 2020 Jan;130:115068. doi.org/10.1016/j.bone.2019.115068
- Ricci KW, Hammill AM, Mobberley-Schuman P, et al. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham-Stout disease. Pediatr Blood Cancer. 2019;66(5):e27614.