




la Newsletter

Michele De Canio, Miriam Teoli, Luca Barbieri, Marco Ardigò
Centro Porfirie e Malattie Rare, Istituto Dermatologico San Gallicano-IRCCS, Roma
Trentacinque anni di follow-up in un paziente affetto da porfiria...
Nel 1987 è stato descritto uno dei pochi casi tuttora noti di...
 Nel novembre 1987 è stato descritto uno dei pochi casi tuttora conosciuti di porfiria epatoeritropoietica (HEP) (1), una forma particolarmente rara di porfiria, gruppo di malattie metaboliche ereditarie correlate alla biosintesi dell’eme (2). Il probando era un bambino di 10 anni che presentava una grave forma di fotosensibilità con comparsa di vescicole sierose, erosioni e croste sul volto e sulle mani (Fig. 1).
Nel novembre 1987 è stato descritto uno dei pochi casi tuttora conosciuti di porfiria epatoeritropoietica (HEP) (1), una forma particolarmente rara di porfiria, gruppo di malattie metaboliche ereditarie correlate alla biosintesi dell’eme (2). Il probando era un bambino di 10 anni che presentava una grave forma di fotosensibilità con comparsa di vescicole sierose, erosioni e croste sul volto e sulle mani (Fig. 1).
La sintomatologia era insorta nei primi anni di vita e durante l’infanzia aveva sviluppato una pronunciata ipertricosi, tuttavia non presentava ritardo nello sviluppo fisico o mentale. Il paziente presentava urine colore rosso pur non avendo anemia emolitica e splenomegalia né eritrodonzia, manifestazioni tipiche della più grave porfiria eritropoietica congenita (CEP; Morbo di Günther). Gli esami ematochimici erano nella norma, ad eccezione dei test di funzionalità epatica che mostravano l’incremento delle transaminasi e un debole aumento della gamma-globulina sierica.
Diagnosi
La diagnosi di HEP è stata stabilita in base al riscontro dell’aumentata escrezione urinaria di porfirine (55% uroporfirina; 28% eptacarbossiporfirina) e dell’incremento di protoporfirina eritrocitaria prevalentemente nella forma legata allo zinco. Negli eritrociti l’attività dell’enzima uroporfirinogeno decarbossilasi (UROD) risultava drasticamente ridotta a circa il 7% del valore normale (1). Successivamente, l’indagine molecolare del gene UROD rivelava la presenza in omozigosi della variante patogenetica c.499G>A [p.Glu167Lys] (3). I genitori, non consanguinei, erano entrambi portatori della mutazione in eterozigosi.
Decorso clinico
Nel corso degli anni il paziente è stato sottoposto a controlli periodici del metabolismo dell’eme, della condizione cutanea ed epatica.
 In assenza di una terapia specifica per la malattia, il rischio di incorrere nei danni cronici dovuti alla fototossicità delle porfirine è stato contenuto tramite l’adozione di misure di prevenzione volte ad evitare l’esposizione alla luce diretta e ai fattori che aggravano la sintomatologia tra cui fumo e l’assunzione di alcol e farmaci inducenti l’attività dei citocromi.
In assenza di una terapia specifica per la malattia, il rischio di incorrere nei danni cronici dovuti alla fototossicità delle porfirine è stato contenuto tramite l’adozione di misure di prevenzione volte ad evitare l’esposizione alla luce diretta e ai fattori che aggravano la sintomatologia tra cui fumo e l’assunzione di alcol e farmaci inducenti l’attività dei citocromi.
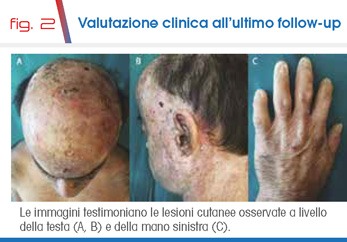
Malgrado il regime fotoprotettivo rigoroso, attualmente, all’età di 46 anni, il paziente mostra numerose lesioni cutanee a diverso stadio di guarigione, esiti cicatriziali e discromie della testa e del collo (Fig. 2 A, B).
Le palpebre mostrano segni di infiammazione cronica con spiccato ectropion in assenza di danni corneali alla valutazione oculistica. Le braccia e le mani presentano segni indicativi di danno fino alla atrofia cutanea simil-sclerodermia con riduzione delle falangi, deformazione articolare, perdita delle unghie (Fig. 2 C). Pur mostrando un severo danno di tipo attinico sulla cute, il paziente non presenta neoplasie alla valutazione in dermatoscopia. Le valutazioni strumentali (eco addominale, RMN epato-pancreatica) evidenziano fibrosi e steatosi epatica ma non epato-splenomegalia. Gli esami ematochimici indicano incremento delle transaminasi e della bilirubina, iperferritinemia come espressione della malattia cronica infiammatoria e del sovraccarico di ferro a livello epatico conseguente all’alterazione metabolica dell’eme. Inoltre si evidenziano segni di anemia emolitica ipercromica e carenza di 25-OH-vitamina D, dovuta probabilmente alla ridotta esposizione al sole.
Discussione
La HEP (MIM #176100) è un raro disordine metabolico congenito della via biosintetica dell’eme, causato da un severo deficit dell’enzima UROD (2). L’accumulo del substrato, nella forma ossidata di uroporfirina, danneggia primariamente epatociti ed eritroblasti, le cellule più attive nella produzione di eme, ma le porfirine rilasciate nel sangue raggiungono la pelle e gli altri organi causando un grave danno ossidativo. Le manifestazioni primarie appaiono precocemente nell’infanzia e comprendono estrema fotosensibilità, fragilità cutanea (vescicole, bolle, erosioni, cicatrici) nelle aree fotoesposte, ipertricosi, eritrodonzia e l’emissione di urine rosso-bruno. Le vescicole tendono a rompersi e guarire lentamente e, nel tempo, ripetute esposizioni alla luce causano cambiamenti sclerodermici che possono evolvere verso la fotomutilazione (4).
Dalla prima descrizione della HEP nel 1969 (5), meno di 100 casi sono stati riportati in letteratura (6). L’HEP è la forma autosomica recessiva della porfiria cutanea tarda familiare (PCT), condizione che predispone alla comparsa della sintomatologia in presenza di fattori di suscettibilità quali epatite C, infezione da HIV, eccessivo consumo di alcol, tabagismo, emocromatosi ereditaria (7). Rispetto alla PCT, le manifestazioni cutanee hanno un esordio precoce, una severità maggiore nella HEP e pertanto appaiono più simili a quelle indotte dalla CEP. Le manifestazioni extracutanee (anemia emolitica, trombocitopenia, splenomegalia, osteolisi, ritardo dello sviluppo) sono invece più frequenti e severe nella CEP che nella HEP (5). La diagnosi differenziale deve considerare PCT, CEP e le altre porfirie ad esordio infantile. L’esame istologico della cute non è utile ai fini diagnostici ma solo un’attenta indagine biochimica può distinguere tra queste patologie che mostrano un quadro clinico sovrapponibile (8).
Conclusioni
Attualmente, non esistono terapie efficaci per ristabilire l’attività enzimatica di UROD negli individui con HEP. La diagnosi precoce, nei centri specializzati, permette di adottare le misure di prevenzione necessarie a contenere l’evoluzione verso il più grave danno cronico. Tuttavia la scarsa conoscenza su questa malattia estremamente rara è spesso causa di ritardo diagnostico con gravi ripercussioni sulla salute del paziente.
Bibliografia
- Bundino S, Topi GC, Zina AM, D'Allessandro Gandolfo L. Hepatoerythropoietic porphyria. Pediatr Dermatol. 1987; 4(3):229-33.
- Stölzel U, Doss MO, Schuppan D. Clinical Guide and Update on Porphyrias. Gastroenterology. 2019; 157(2):365-381.e4.
- Romana M, Grandchamp B, Dubart A, et al. Identification of a new mutation responsible for hepatoerythropoietic porphyria. Eur J Clin Invest. 1991; 21(2):225–229.
- Elder GH. Hepatic porphyrias in children. J Inher Metab Dis. 1997; 20(2):237–246.
- Piñol Aguadé J, Castells A, Indacochea A, Rodés J. A case of biochemically unclassifiable hepatic porphyria. Br J Dermatol. 1969; 81(4):270–275.
- Liu LU, Phillips J, Bonkovsky H, et al. Hepatoerythropoietic Porphyria. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews®. Seattle, WA: University of Washington, 1993. Available at http://www.ncbi.nlm.nih.go/books/NBK169003/.
- Egger NG, Goeger DE, Payne DA, et al. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci. 2002; 47:419–426.
- Phillips JD. Heme biosynthesis and the porphyrias. Mol Genet Metab. 2019; 128(3):164-177.