




la Newsletter
Una condizione ultra rara da scoprire: la sindrome di Kleefstra |...
Inizialmente descritta come “sindrome da delezione 9q...
V. è un bambino di 3 anni e 11 mesi che conosciamo dai 15 mesi perché inviato per una visita genetica di inquadramento. Il piccolo è nato alla 38° settimana di EG da parto eutocico dopo gravidanza normodecorsa. Il peso alla nascita era 2,600 kg (97°p.le), l'APGAR 9/10. Riferito buon adattamento alla vita extra-uterina. La crescita staturo-ponderale è riferita costante, ai centili superiori della norma. In anamnesi vengono riportati tre episodi di convulsione febbrile semplice ed un ricovero a 15 mesi per polmonite dispnoizzante oltre a svariati episodi infettivi. La visita oculistica mostra un’escavazione della papilla (pseudocoloboma). La RMN evidenzia una modesta ipoplasia del corpo calloso.
 Al momento della valutazione V. frequenta la scuola dell’infanzia con sostegno per un quadro di moderato ritardo psicomotorio; è presente un modesto ipertono a carico degli arti inferiori ed una discreta scialorrea.
Al momento della valutazione V. frequenta la scuola dell’infanzia con sostegno per un quadro di moderato ritardo psicomotorio; è presente un modesto ipertono a carico degli arti inferiori ed una discreta scialorrea.
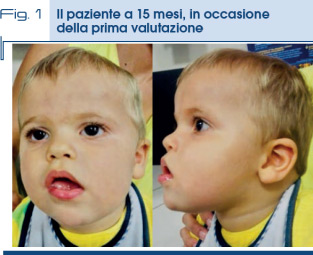
All’età dell’ultima valutazione pronuncia qualche parola con buon intento comunicativo. All’esame obiettivo sono presenti sfumate anomalie minori del viso tra cui la presenza di occhi infossati, labbra carnose con labbro superiore incurvato, denti spaziati ed una ipoplasia medio facciale (Fig. 1). Ha eseguito un arrayCGh risultato nella norma. L’insieme dei dati clinici è comunque orientativo per un quadro complesso su base costituzionale. In assenza di diagnosi cliniche gestaltiche il piccolo viene sottoposto a Whole Exome Sequencing che non mostra varianti patogenetiche significative.
La rivalutazione del quadro con l’utilizzo del programma Face2gene suggerisce un’ipotesi di sindrome di Kleefstra. Poiché tale ipotesi sembra convincente andiamo ad analizzare perché i test genetici sin qui eseguiti non siano stati di aiuto. Questo approfondimento ci permette di capire che una quota minoritaria di questi pazienti può avere una delezione intragenica a carico del gene malattia EHMT1.
Attiviamo, quindi, una indagine MLPA del gene stesso che ci permette di mettere in luce una delezione dell’esone 19 che conferma la diagnosi clinica.
La sindrome di Kleefstra (KS)
Inizialmente descritta come “sindrome da delezione 9q subtelomerica”, è una rara malattia genetica caratterizzata da dismorfismi, ipotonia, disabilità intellettiva con grave ritardo del linguaggio (1) descritta la prima volta nel 1999; dal 2010 la condizione ha preso il nome della dottoressa olandese Tjitske Kleefstra che si dedica attivamente allo studio di questa sindrome.
Ad oggi sono descritti circa 100 pazienti con diagnosi certa (2), ma la prevalenza può essere considerata più alta tenuto conto delle probabili mancate diagnosi (3); maschi e femmine sono affetti in proporzione similare.
Il difetto di base è rappresentato nel 50-80% dei casi da una microdelezione della regione cromosomica 9q34.3 e nel restante 20-50% da mutazioni/anomalie intrageniche del gene EHMT1 (Euchromatin Histone Methyl-Transferase 1) mappato all’interno della regione cromosomica 9q34.3 stessa (3) (4). Il gene codifica per una proteina di 1298 aminoacidi coinvolta nel processo di metilazione del DNA.
La diagnosi
La diagnosi di KS fondata sul sospetto clinico può trovare conferma biologica attraverso arrayCGH per l’identificazione di duplicazioni/delezioni della regione cromosomica 9q34.3 o sequenziamento del gene EHMT1 (analisi diretta, WES, pannelli genomici per disabilità intellettiva contenenti il gene in oggetto). È necessaria l’analisi MLPA per l’identificazione di piccole delezioni intrageniche descritte nel 5% dei pazienti (5). Le anomalie genetiche sopracitate sono solitamente ad insorgenza casuale, de novo (90%), nel soggetto affetto; è, quindi, rara l’osservazione di una ricorrenza familiare. La presenza di un mosaicismo somatico (o germinale) per uno dei difetti o di una traslocazione cromosomica bilanciata coinvolgente la regione 9q34.3 in uno dei genitori rappresentano elementi favorenti una ricorrenza familiare della patologia.
Caratteristiche cliniche
Sul piano fenotipico i bambini affetti presentano anomalie minori del viso abbastanza specifiche quali: brachi-microcefalia, fronte ampia, sopracciglia arcuate, sinofria, lieve up slanting delle rime palpebrali, elice ispessito, naso corto con narici anteverse, ipoplasia mediofacciale, labbro superiore incurvato, labbro inferiore extroverso, lingua protrusa e prognatismo (5). I tratti del viso si fanno più accentuati con il passare degli anni. Frequenti sono inoltre le anomalie dentarie (presenza di denti neonatali o ritardata perdita della dentizione primaria).
Il quadro tipico della KS si caratterizza inoltre con:
Accrescimento staturo ponderale: il peso alla nascita dei pazienti con KS è normale o ai limiti superiori di norma (10% dei casi delezione 9q34.3 e 50% mutazione EHMT1) ed evolve, in circa la metà dei casi, verso quadri di sovrappeso e obesità. La statura è solitamente nei range di normalità.
Ritardo psicomotorio/disabilità intellettiva: è comune il riscontro di ipotonia nei primi mesi/anni di vita con conseguente ritardo nell’acquisizione delle normali tappe di sviluppo motorio. La deambulazione autonoma può avvenire intorno ai 2-3 anni (3). I pazienti mostrano usualmente quadro di disabilità intellettiva moderata/severa; sono però descritti pazienti con un ritardo mentale lieve o QI ai limiti inferiori di norma nell’ambito di un disturbo dello spettro autistico (1).
Data la non elevata numerosità dei soggetti noti, non è ancora definibile in modo assoluto la reale variabilità di espressione clinica. È descritta la presenza di un grave ritardo del linguaggio espressivo, mentre risulta maggiormente preservata la comprensione del linguaggio nell’ambito di una cosiddetta “childhood apraxia of speech”. In relazione a ciò alcuni pazienti utilizzano con buoni risultati sistemi di comunicazione non verbale (2).
Ulteriori problematiche relative alla sfera del neurosviluppo e del comportamento sono rappresentate da disturbi dello spettro autistico, stereotipie e modesti atteggiamenti di autoaggressività che a volte evolvono in quadri di apatia e catatonia nell’adulto.
Malformazioni maggiori: non esiste una malformazione maggiore tipica della condizione ed evocativa della stessa come avviene in altre condizioni sindromiche.
Sono segnalati con una prevalenza significativamente maggiore rispetto alla popolazione generale i seguenti difetti congeniti: anomalie cardiache osservate nel 30% dei casi ed estremamente variabili in termini di tipologia (difetti del setto atriale e ventricolare, tetralogia di Fallot, coartazione aortica, valvola aortica bicuspide e stenosi polmonare); alterazioni del sistema nervoso centrale (ipoplasia del corpo calloso, anomalie corticali e difetti della sostanza bianca); anomalie renali (idronefrosi, cisti renali, reflusso vescico-ureterale); anomalie dei genitali nei maschi (ipospadia, criptorchidismo e micropene).
Comorbidità pediatriche: i pazienti con KS mostrano non raramente problematiche gastro-intestinali quali reflusso gastroesofageo, addominalgia, stipsi. Sono occasionalmente stati osservati bambini con infezioni respiratorie ricorrenti. È inoltre possibile l’insorgenza di episodi critici convulsivi (30% dei casi con delezione 9q34.3 e 20% dei casi con mutazione EHMT1) e quadri epilettici (crisi tonico cloniche, assenze, epilessia parziale complessa) (2).
In ambito sensoriale alcuni pazienti presentano ipoacusia neurosensoriale o trasmissiva ed alterazioni dell’acuità visiva (ipermetropia). Una potenziale complicanza cardiologica non malformativa è rappresentata dalla presenza di flutter atriale. I pazienti con KS possono presentare disturbi del sonno con frequenti risvegli notturni.
Alcuni report segnalano che nei pazienti adolescenti o giovani adulti i disturbi del sonno possono anticipare la comparsa di una regressione con sviluppo di psicosi, depressione o disturbi bipolari. Un precoce trattamento farmacologico con antipsicotici è fondamentale per migliorare l’outcome a lungo termine rallentando l’evolutività del quadro psichico (6).
In termini di correlazione genotipo-fenotipo i dati ad oggi disponibili sembrano evidenziare come i quadri secondari a microdelezione (se di ampiezza >1 Mb) si associno a fenotipi più severi (4) rispetto a quelli in cui il difetto di base è rappresentato da microdelezione di ampiezza inferiore ad 1Mb o a mutazioni intrageniche, sia in termini di gravità della disabilità intellettiva che di frequenza/severità delle comorbidità mediche (3).
 La KS entra in diagnosi differenziale con numerose condizioni sindromiche quali la sindrome di Down, di Smith-Magenis, di Pitt-Hopkins, di Angelman, la sindrome KMT2C associata e l’aploinsufficienza di MBD5. La tabella 1 descrive gli elementi clinici di overlapping tra la KS e le sindromi citate (4).
La KS entra in diagnosi differenziale con numerose condizioni sindromiche quali la sindrome di Down, di Smith-Magenis, di Pitt-Hopkins, di Angelman, la sindrome KMT2C associata e l’aploinsufficienza di MBD5. La tabella 1 descrive gli elementi clinici di overlapping tra la KS e le sindromi citate (4).
Conclusioni e prospettive
Come intuibile c’è ancora molto lavoro da fare per la definizione accurata sia della variabilità di espressione clinica della condizione sia della sua reale storia naturale. In analogia a tutte le condizioni sindromiche multisistemiche il followup assistenziale si giova di un percorso integrato tra un ambito neurospichiatrico/riabilitativo (la cui efficacia rappresenta l’elemento maggiormente determinante la prognosi del paziente) ed un ambito internistico (inizialmente pediatrico) per la diagnosi precoce ed il trattamento delle potenziali complicanze mediche note (gastrointestinali, respiratorie, cardiache, neurosensoriali, neurologiche e del sonno).
A ottobre 2019 è nata l'Associazione Italiana Sindrome di Kleefstra APS - Tel: 347-8318440 - www.kleefstraitalia.org - e-mail: kleefstraitalia@gmail.com.
Bibliografia
- Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, et al. Update on Kleefstra Syndrome. Mol Syndromol. 2012; 2(3-5):202–212.
- Samango-Sprouse C, Lawson P, Spouse C, et al. Expanding the Phenotypic profile of Kleefstra Syndrome: A Female with Low-Avarage Intelligence and Childhood Apraxia of Speech. Am J Med Genet Part A. 2016;170A:1312-1316.
- Kleefstra T, de Leeuw N. Kleefstra Syndrome. Gene Reviews 2010 Oct 5 (Updated 2019 Mar 21).
- Ciaccio C, Scuvera G, Tucci A, et al. New Insights into Kleefstra Syndrome: Report of Two Novel Cases with Previosly Unreported Features and Literature Review. Cytogenet Genome Res. 2018;156(3):127-133.
- https://www.orpha.net/consor4.01/www/cgi-bin/?lng=IT
- Vermeulen K, Staal WG, Janzing JG, et al. Sleep Disturbance as a Precursor of Severe Regression in Kleefstra Syndrome Suggests a Need for Firm and Rapid Pharmacological Treatment. Clin Neuropharm. 2017;40(4):185-188.