




la Newsletter
Una forma atipica di osteogenesi imperfetta: la tipo V | La...
La osteogenesi imperfetta (OI) tipo V è una patologia molto rara...
Z. giunge alla nostra osservazione in ambulatorio di genetica clinica, all’età di 25 anni, con una diagnosi di fibrodisplasia ossificante progressiva (FOP), una malattia gravemente disabilitante caratterizzata da ossificazione eterotopica progressiva in siti extrascheletrici.
Non sono disponibili informazioni sulla storia familiare, perinatale e dei primi anni di vita in quanto adottato all’età di tre anni. Il ragazzo riferisce di aver sempre manifestato difficoltà nella prono-supinazione degli arti superiori.
In seguito ad un trauma, all’età di sette anni, si è verificata una frattura del femore sinistro e in sede di frattura si è sviluppata un’iperplasia ossea, che ha portato al sospetto diagnostico prima di miosite ossificante e in seguito di FOP e a numerosi interventi chirurgici, per il persistere di dolori diffusi e limitazioni funzionali alla colonna vertebrale e agli arti. Nel percorso diagnostico sono stati effettuati diversi esami clinici e strumentali e in questo contesto il paziente è stato sottoposto per tre volte a biopsia del femore sinistro, di cui non sono disponibili i referti. Nel corso del tempo il paziente è stato sottoposto ad altri interventi chirurgici: la resezione del capitello radiale destro per la persistente e ingravescente difficoltà di prono-supinazione e il cerchiaggio della rotula sinistra in seguito a frattura scomposta. Non vengono riferiti altri problemi eccetto una miopia in follow-up.
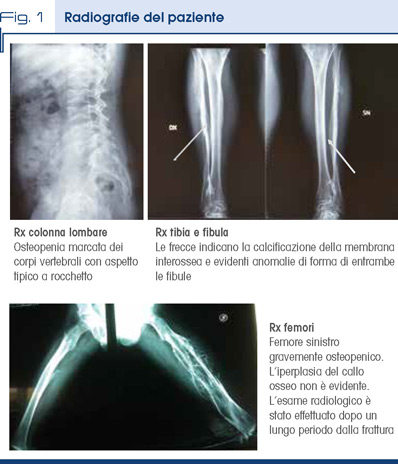 All’esame clinico: peso 70 Kg, altezza 164 cm e circonferenza cranica 57 cm; malocclusione di tipo III, limitata prono-supinazione e flessione degli avambracci più marcata a destra, deviazione radiale dell’indice della mano destra, dismetria degli arti inferiori (sn>dx, 1.5 cm), riduzione della flessione dell’anca sinistra. Le indagini radiografiche esibite mostrano un’iperplasia del callo osseo al femore sinistro, aspetto biconcavo dei corpi vertebrali più evidente in regione lombare, calcificazione della membrana interossea agli arti inferiori (Fig.1); sono assenti sclere blu e dentinogenesi imperfetta. Indagini richieste: profilo biochimico calcio-fosforo (sierico e urinario), fosfatasi alcalina, PTH, osteocalcina e Vitamina D nella norma; osteoporosi conclamata alla MOC; ECG ed ecocardiogramma normali, PA 120/80; audiometria normale.
All’esame clinico: peso 70 Kg, altezza 164 cm e circonferenza cranica 57 cm; malocclusione di tipo III, limitata prono-supinazione e flessione degli avambracci più marcata a destra, deviazione radiale dell’indice della mano destra, dismetria degli arti inferiori (sn>dx, 1.5 cm), riduzione della flessione dell’anca sinistra. Le indagini radiografiche esibite mostrano un’iperplasia del callo osseo al femore sinistro, aspetto biconcavo dei corpi vertebrali più evidente in regione lombare, calcificazione della membrana interossea agli arti inferiori (Fig.1); sono assenti sclere blu e dentinogenesi imperfetta. Indagini richieste: profilo biochimico calcio-fosforo (sierico e urinario), fosfatasi alcalina, PTH, osteocalcina e Vitamina D nella norma; osteoporosi conclamata alla MOC; ECG ed ecocardiogramma normali, PA 120/80; audiometria normale.
L’insieme dei dati ha consentito di porre la diagnosi clinica di osteogenesi imperfetta (OI) tipo V. L’indagine molecolare del gene causativo IFITM5 ha confermato la diagnosi, rilevando la presenza della variante patogenetica c.-14C>T, in eterozigosi.
Osteogenesi imperfetta tipo V
L’OI di tipo V (MIM # 610967) è una forma rara e peculiare (circa 100 casi pari al 4% del totale dei casi di OI). Ha una trasmissione autosomica dominante come l’85% dei casi dovuti ad anomalie del collageno di tipo 1. Le forme recessive sono sempre clinicamente gravi e geneticamente eterogenee (finora dovute ad altri 16 geni). La prevalenza di tutte le forme è pari a 1/13-14000 nati. Il fenotipo clinico della OI V è rappresentato, come nelle altre forme, dalla osteopenia con fragilità ossea e predisposizione alle fratture e dalla bassa statura; la dentinogenesi imperfetta e la presenza di sclere bluastre sono invece rilevabili in un numero estremamente ridotto di casi. I segni radiografici tipici, assenti nelle altre forme di OI, sono: l’iperplasia del callo osseo, la dislocazione del radio, la calcificazione delle membrane interossee tra radio e ulna e tra tibia e fibula e la presenza di linee dense metafisarie. I corpi vertebrali presentano il profilo biconcavo, “a rocchetto”, tipico delle OI tipo III/IV.
Patogenesi
L’OI di tipo V è causata da varianti patogenetiche del gene IFITM5 (Interferon-Induced Transmembrane Protein 5) che codifica la proteina BRIL (Bone Restricted IFITM-like). BRIL è una proteina di membrana degli osteoblasti con due domini transmembrana che facilita la mineralizzazione ossea regolando l’espressione di un gene specifico degli osteoblasti, SERPINF1 che codifica per la proteina PEDF (Pigmented Epithelium-Derived Factor), che facilita la differenziazione e mineralizzazione degli osteoblasti.
Le varianti patogenetiche note del gene IFITM5 predispongono ad un’ossificazione ectopica dovuta a un’iperattivazione del gene SERPINF1.
La mutazione ricorrente nei pazienti con OI di tipo V è la variante patogenetica c.-14C>T, che determina nella regione 5’UTR del gene la presenza di un nuovo sito di inizio (ATG) della sintesi proteica introducendo una sequenza di 5 ulteriori aminoacidi, nota come MALEP (Met-Ala-Leu-Glu-Pro), alla porzione N-terminale della proteina.
Segni clinici
È riportata variabilità fenotipica sia intra- che inter-familiare. Il segno cardine è la calcificazione delle membrane interossee; la bassa statura è costante ma presente con gravità variabile. L’iperplasia del callo osseo è tipica, ma non sempre presente. Sono comuni scoliosi o fratture da compressione dei corpi vertebrali e meno frequenti coxartriti e contratture articolari. Un’altra variante causativa nel gene IFITM5 (c.119C>T, p.Ser40Leu) introduce la sostituzione di una serina con leucina. I sette pazienti con questa mutazione hanno un fenotipo clinico più grave (simil OI tipo VI da varianti patogenetiche di SERPINF1) con fratture multiple ad esordio prenatale, bassa statura grave, macrocefalia, brevità e incurvamento degli arti inferiori e marcata compromissione della deambulazione; non è presente iperplasia del callo osseo.
È riportato un paziente con lo stesso fenotipo ma con una variante di transizione nello stesso codone (c.119C>G, p.Ser40Trp). Infine un’ulteriore variante causa del fenotipo V è dovuta alla variante MEP, (c.-9C>A) che introduce nella zona 5’UTR di IFITM5 tre aminoacidi (metionina, glutammina, prolina).
Conclusioni
I pazienti affetti da OI di tipo V, così come è noto per gli altri pazienti con OI, possono essere trattati con bifosfonati che migliorano l’osteopenia; tuttavia in letteratura sono descritti risultati contrastanti.
L’osteogenesi imperfetta di tipo V è una patologia molto rara con caratteristiche peculiari nel contesto delle OI.
Va sospettata sempre di fronte ad un paziente con bassa statura e fragilità ossea che mostri la presenza di un’iperplasia ossea nella sede di una frattura e la calcificazione della membrana interossea.
Il corretto inquadramento clinico consente una diagnosi etiologica rapida evitando ritardi nella diagnosi e approcci errati, un corretto programma di follow-up e di terapia, nonché una valutazione del rischio di ricorrenza familiare.
Bibliografia
- Semler O, Garbes L, Keupp K et al. A Mutation in the 50-UTR of IFITM5 Creates an In-Frame Start Codon and Causes Autosomal-Dominant Osteogenesis Imperfecta Type V with Hyperplastic Callus. Am J Hum Genet 2012; 91:349-357.
- Shapiro JR, Lietman CD, Glover M et al. Phenotypic Variability of Osteogenesis Imperfecta Type V Caused by an IFITM5 Mutation. J Bone Miner Res 2013; 28:1523-1530.
- Rodriguez Celin M, Moosa S, Fano V. Uncommon IFITM5 mutation associated with severe skeletal deformity in osteogenesis Imperfecta. Ann Hum Genet 2018; 82:477-481.
- Lim JY, Bhatia NS, Vasanwala RS. A novel Ser40Trp variant in IFITM5 in a family with osteogenesis imperfecta and review of the literature. Clin Dysmorphol 2019; 28:118–123.
- Cao YJ, Wei Z, Zhang H and Zhang ZL. Expanding the Clinical Spectrum of Osteogenesis Imperfecta Type V: 13 Additional Patients and Review. Front Endocrinol 2019; 10:375.
- Besio R, Chow CW, Tonelli F, Marini JC, and Forlino A. Bone biology: insights from osteogenesis imperfecta and related rare fragility syndromes. The FEBS Journal 2019, 286: 3033–3056